

Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing
- PMID: 30116051
- PMCID: PMC6456907
- DOI: 10.1038/nrd.2018.109
Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing
Abstract
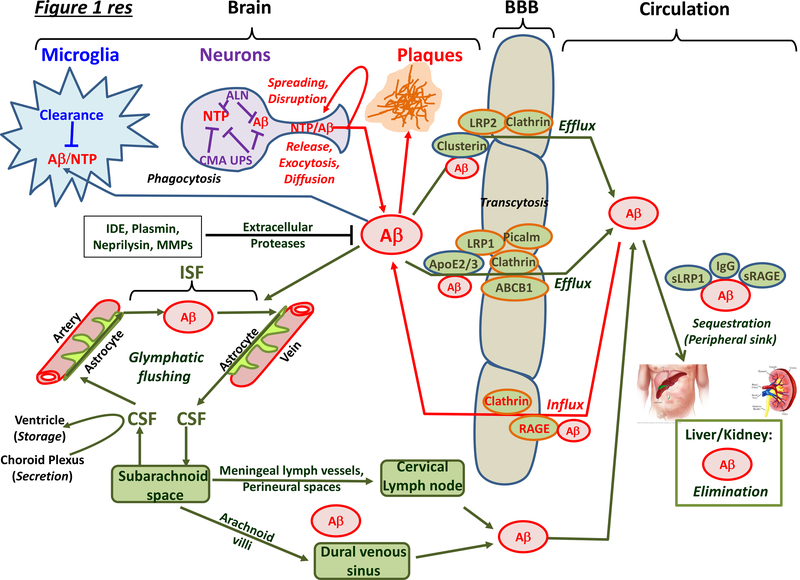
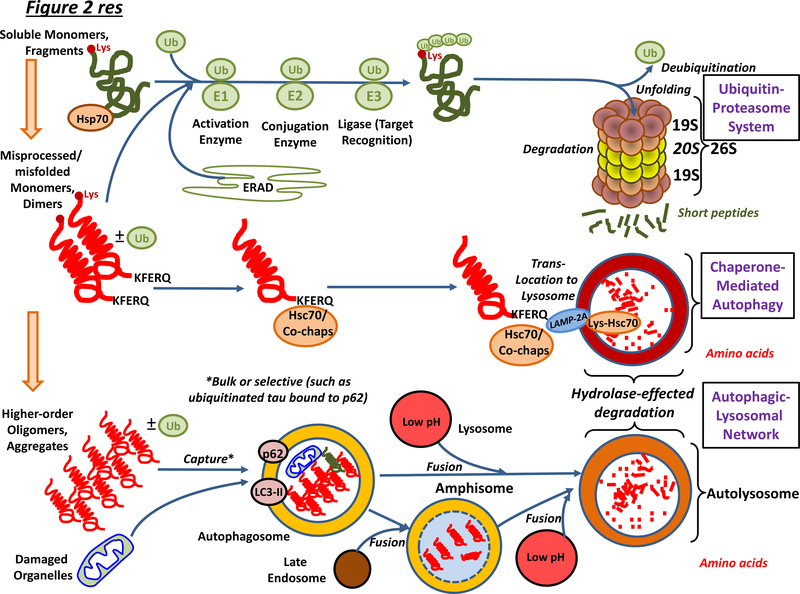
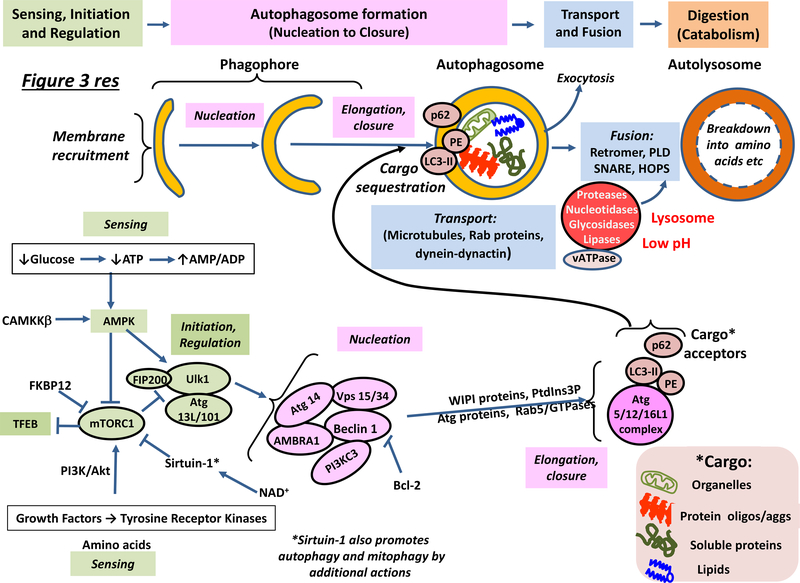
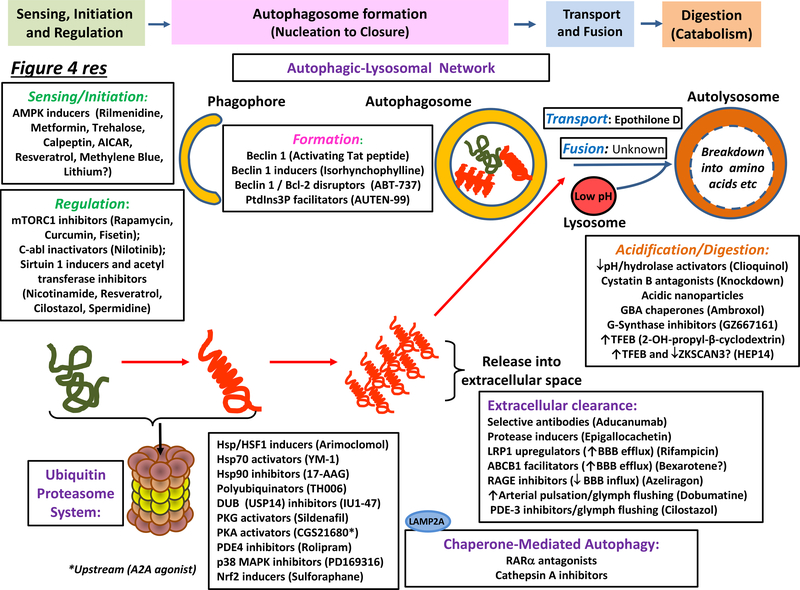
Neurodegenerative disorders of ageing (NDAs) such as Alzheimer disease, Parkinson disease, frontotemporal dementia, Huntington disease and amyotrophic lateral sclerosis represent a major socio-economic challenge in view of their high prevalence yet poor treatment. They are often called 'proteinopathies' owing to the presence of misfolded and aggregated proteins that lose their physiological roles and acquire neurotoxic properties. One reason underlying the accumulation and spread of oligomeric forms of neurotoxic proteins is insufficient clearance by the autophagic-lysosomal network. Several other clearance pathways are also compromised in NDAs: chaperone-mediated autophagy, the ubiquitin-proteasome system, extracellular clearance by proteases and extrusion into the circulation via the blood-brain barrier and glymphatic system. This article focuses on emerging mechanisms for promoting the clearance of neurotoxic proteins, a strategy that may curtail the onset and slow the progression of NDAs.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
