

Identification of Positively and Negatively Selected Driver Gene Mutations Associated With Colorectal Cancer With Microsatellite Instability
- PMID: 30116770
- PMCID: PMC6089198
- DOI: 10.1016/j.jcmgh.2018.06.002
Identification of Positively and Negatively Selected Driver Gene Mutations Associated With Colorectal Cancer With Microsatellite Instability
Abstract
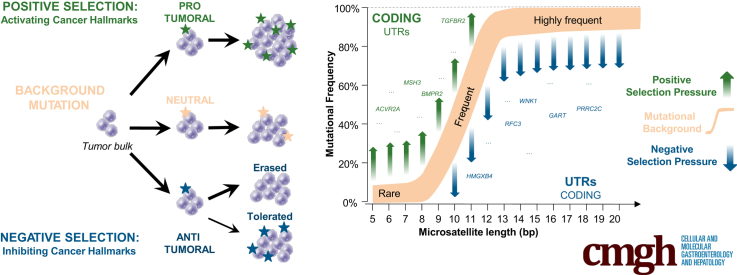
Background & aims: Recent studies have shown that cancers arise as a result of the positive selection of driver somatic events in tumor DNA, with negative selection playing only a minor role, if any. However, these investigations were concerned with alterations at nonrepetitive sequences and did not take into account mutations in repetitive sequences that have very high pathophysiological relevance in the tumors showing microsatellite instability (MSI) resulting from mismatch repair deficiency investigated in the present study.
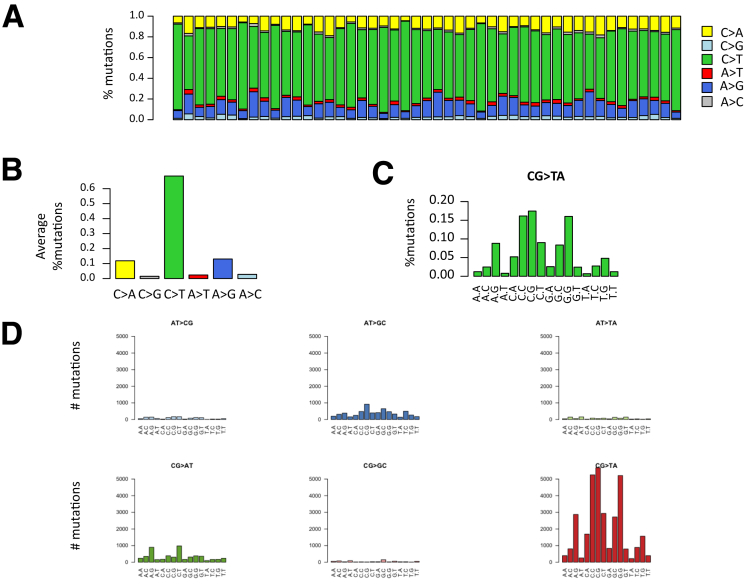
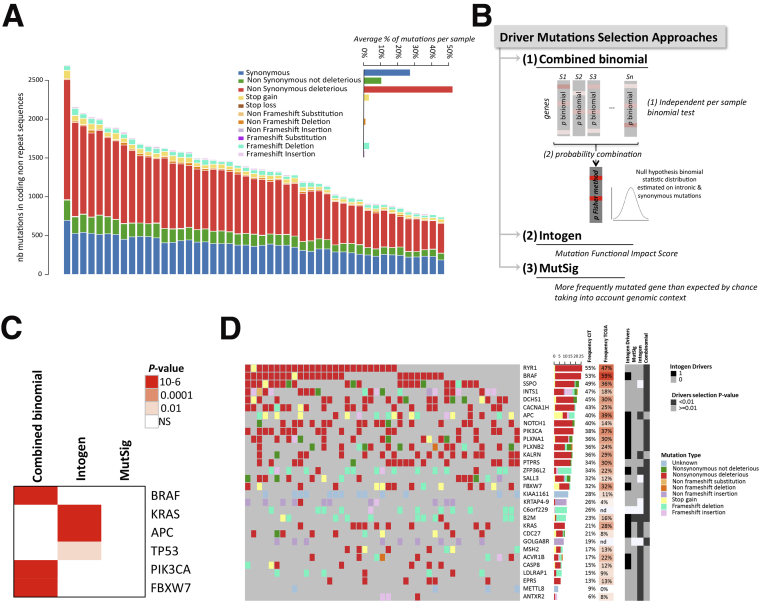
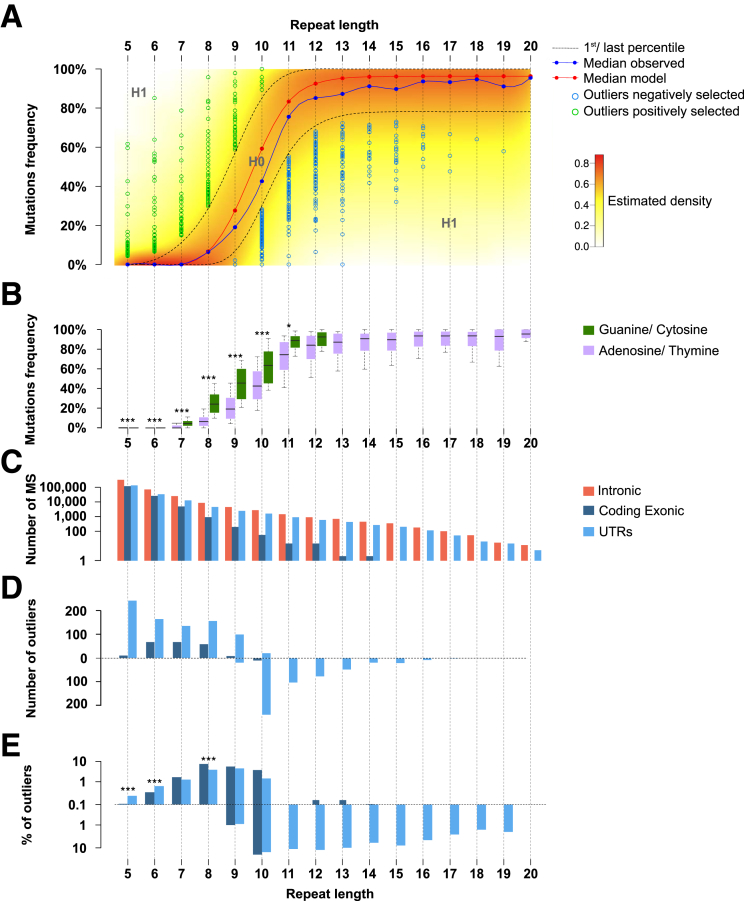
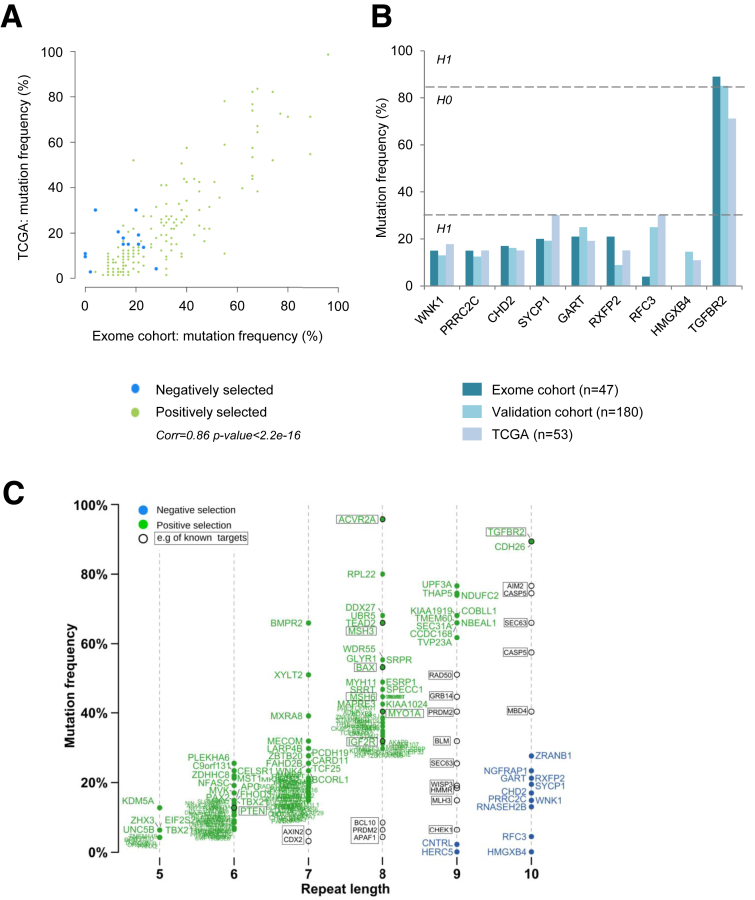
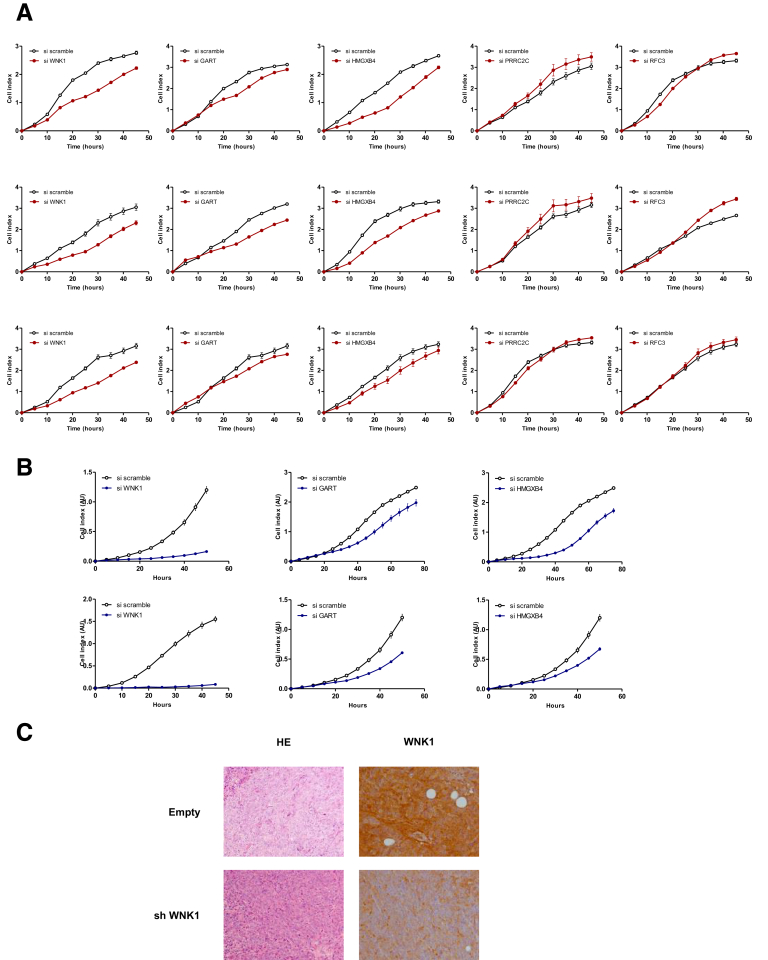
Methods: We performed whole-exome sequencing of 47 MSI colorectal cancers (CRCs) and confirmed results in an independent cohort of 53 MSI CRCs. We used a probabilistic model of mutational events within microsatellites, while adapting pre-existing models to analyze nonrepetitive DNA sequences. Negatively selected coding alterations in MSI CRCs were investigated for their functional and clinical impact in CRC cell lines and in a third cohort of 164 MSI CRC patients.
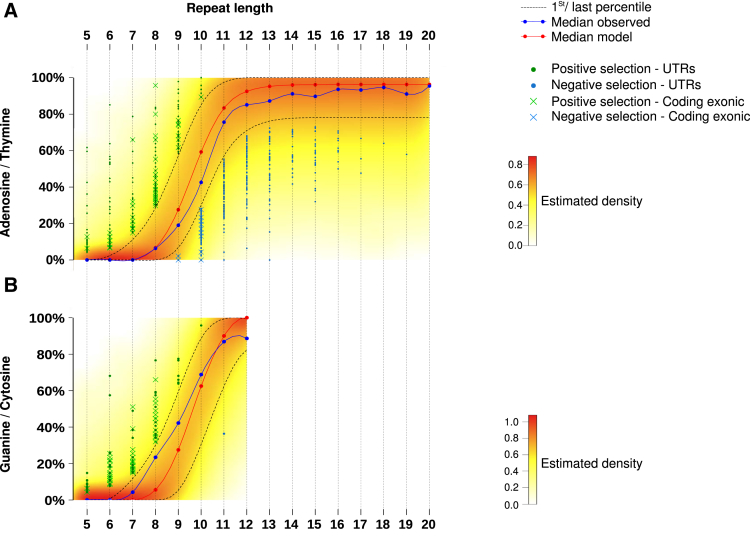
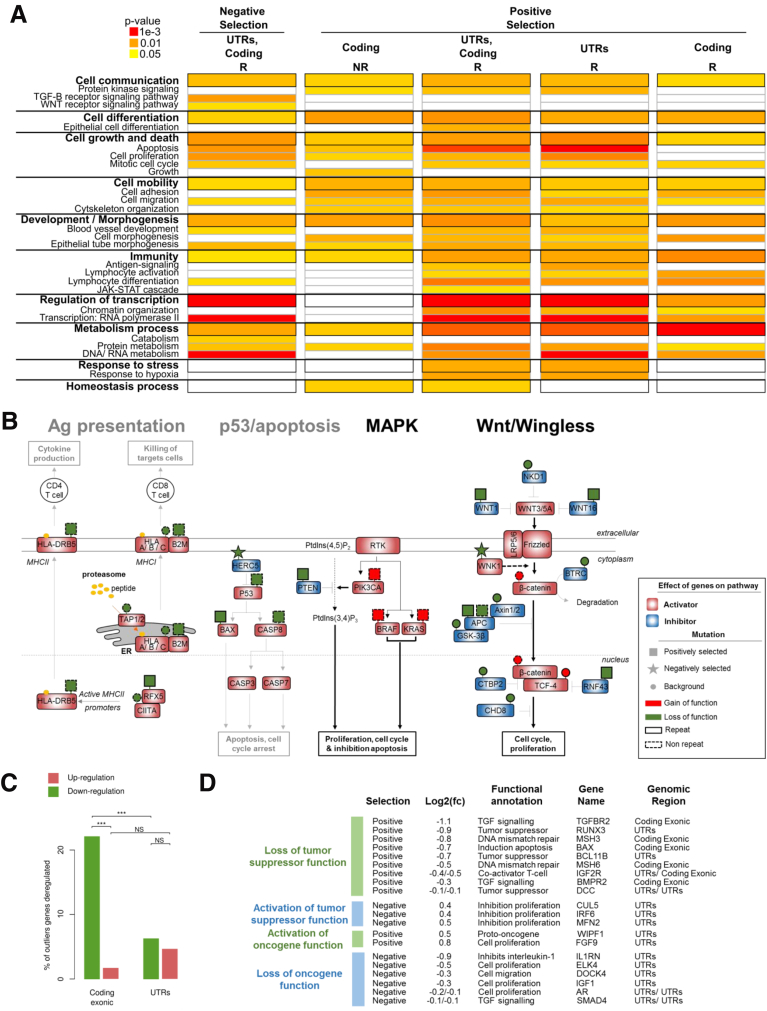
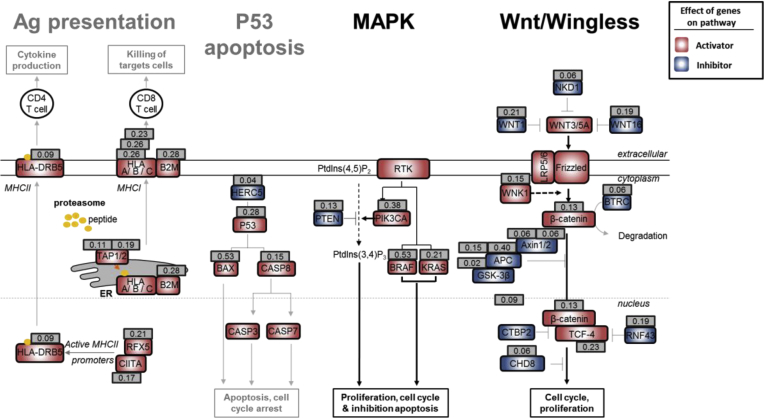
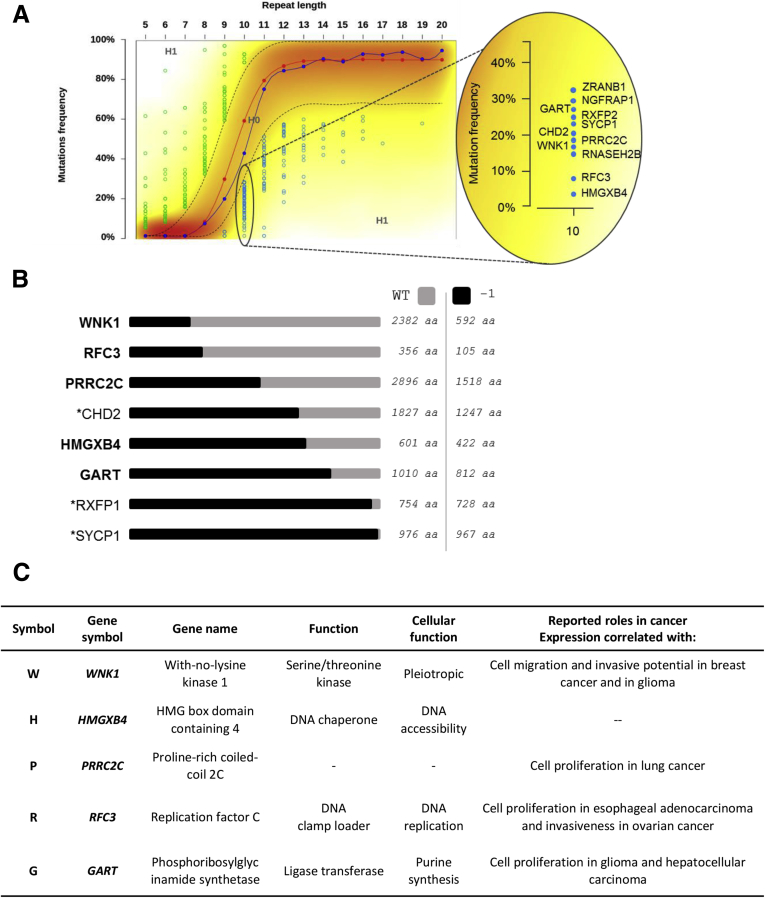
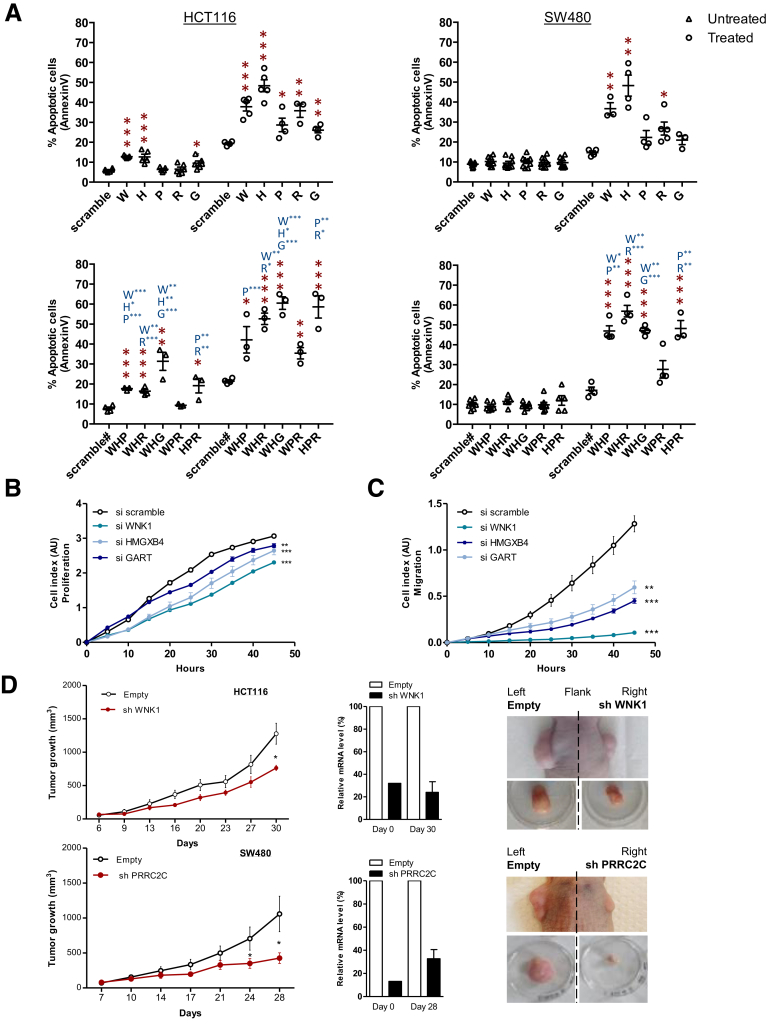
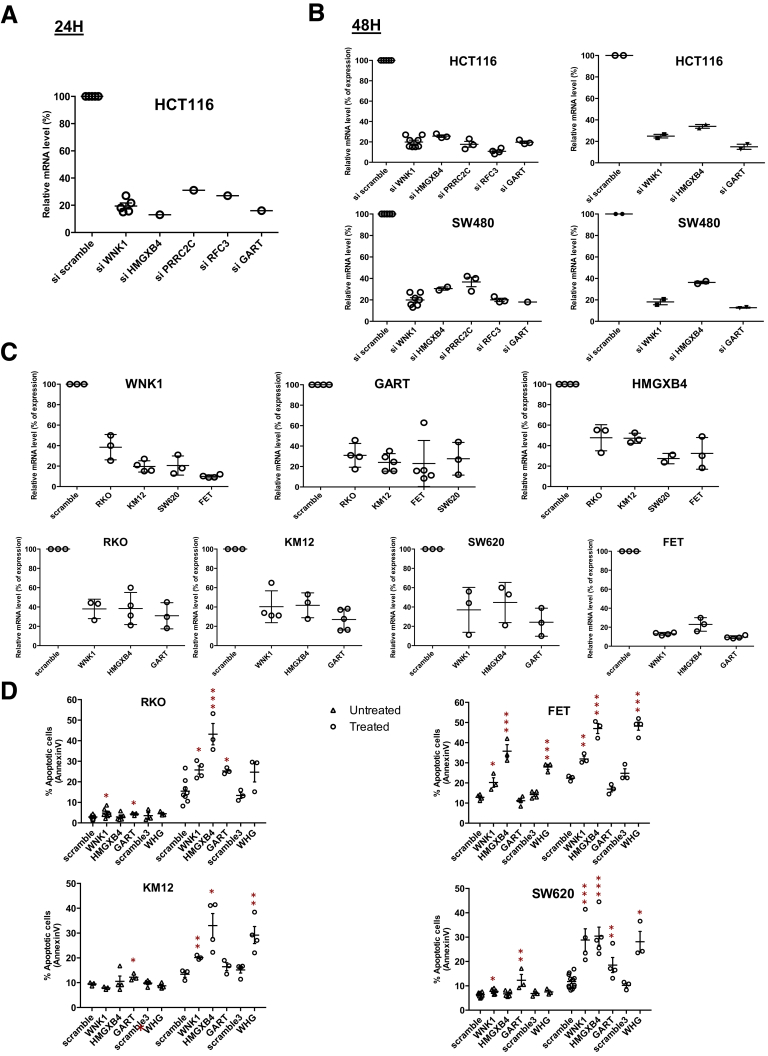
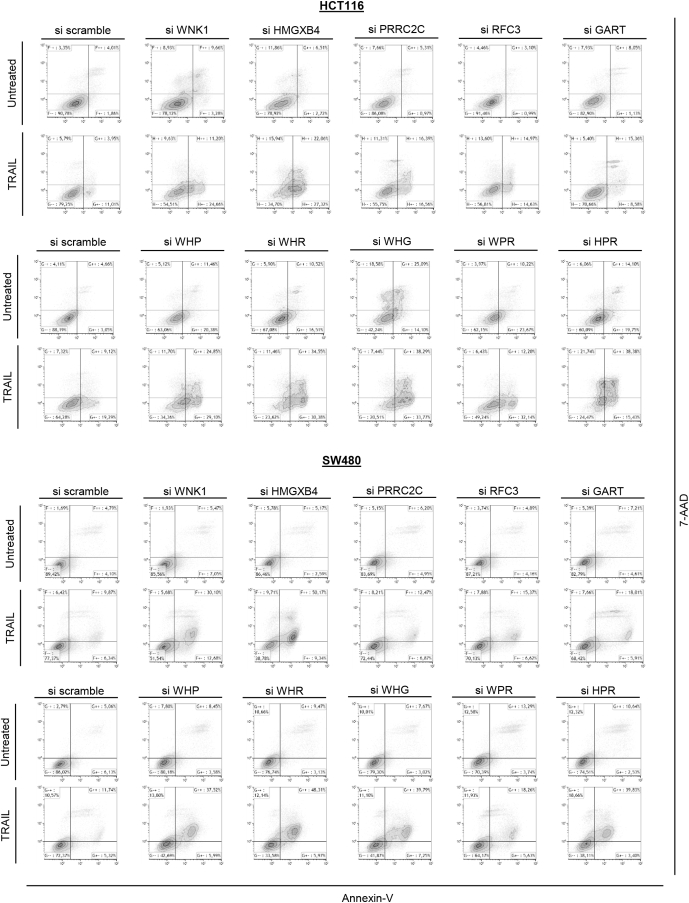
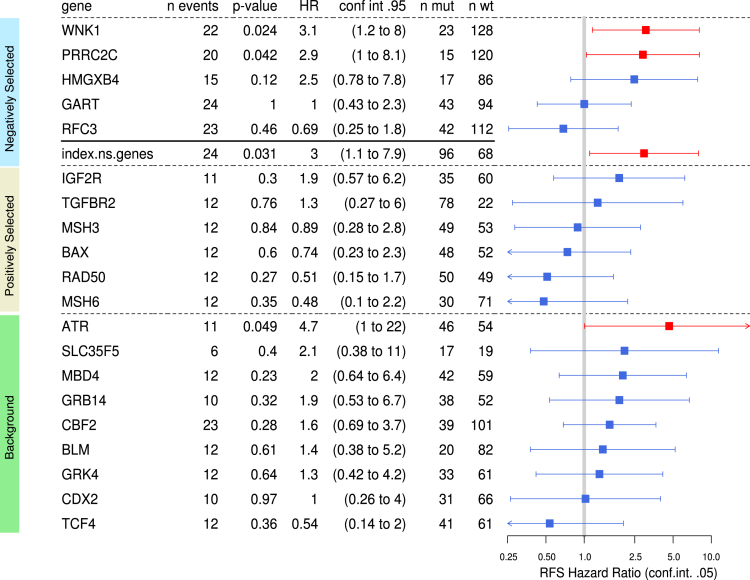
Results: Both positive and negative selection of somatic mutations in DNA repeats was observed, leading us to identify the expected true driver genes associated with the MSI-driven tumorigenic process. Several coding negatively selected MSI-related mutational events (n = 5) were shown to have deleterious effects on tumor cells. In the tumors in which deleterious MSI mutations were observed despite the negative selection, they were associated with worse survival in MSI CRC patients (hazard ratio, 3; 95% CI, 1.1-7.9; P = .03), suggesting their anticancer impact should be offset by other as yet unknown oncogenic processes that contribute to a poor prognosis.
Conclusions: The present results identify the positive and negative driver somatic mutations acting in MSI-driven tumorigenesis, suggesting that genomic instability in MSI CRC plays a dual role in achieving tumor cell transformation. Exome sequencing data have been deposited in the European genome-phenome archive (accession: EGAS00001002477).
Keywords: CRC, colorectal cancer; Colorectal Cancer; Driver Gene Mutations; HR, hazard ratio; MLH1, MutL Homolog 1; MMR, mismatch repair; MSH, MutS Homolog; MSI, microsatellite instability; Microsatellite Instability; NR, nonrepetitive; PBS, phosphate-buffered saline; PCR, polymerase chain reaction; Positive and Negative Selection; R, repetitive; RFS, relapse-free survival; RTCA, Real-Time Cell Analyzer; Tumorigenic Process; UTR, untranslated region; WES, whole-exome sequencing; WGA, whole-genome amplification; bp, base pair; indel, insertion/deletion; mRNA, messenger RNA; shRNA, short hairpin RNA; siRNA, small interfering RNA.
Figures

Comment in
-
Colon Cancers Get a Negative (Selection) Attitude.Cell Mol Gastroenterol Hepatol. 2018 Jul 20;6(3):349. doi: 10.1016/j.jcmgh.2018.06.009. eCollection 2018. Cell Mol Gastroenterol Hepatol. 2018. PMID: 30182045 Free PMC article. No abstract available.
References
-
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
-
- Bakhoum S.F., Landau D.A. Cancer evolution: no room for negative selection. Cell. 2017;171:987–989. - PubMed
-
- Leach F.S., Nicolaides N.C., Papadopoulos N., Liu B., Jen J., Parsons R., Peltomaki P., Sistonen P., Aaltonen L.A., Nystrom-Lahti M., Zhang G.J., Meltzer P.S., Yu J.W., Kao F.T., Chen D.J., Cerosaletti K.M., Fournier R.E.K., Todd S., Lewis T., Leach R.J., Naylor S.L., Weissenbach J., Mecklin J.P., Jarvinen H., Petersen G.M., Hamilton S.R., Green J., Jass J., Watson P., Lynch H.T., Trent J.M., de la Chapelle A., Kinzler K.W., Vogelstein B. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
