

SUMOylation of VEGFR2 regulates its intracellular trafficking and pathological angiogenesis
- PMID: 30120232
- PMCID: PMC6098000
- DOI: 10.1038/s41467-018-05812-2
SUMOylation of VEGFR2 regulates its intracellular trafficking and pathological angiogenesis
Erratum in
-
Author Correction: SUMOylation of VEGFR2 regulates its intracellular trafficking and pathological angiogenesis.Nat Commun. 2019 Aug 15;10(1):3679. doi: 10.1038/s41467-019-11659-y. Nat Commun. 2019. PMID: 31417083 Free PMC article.
Abstract
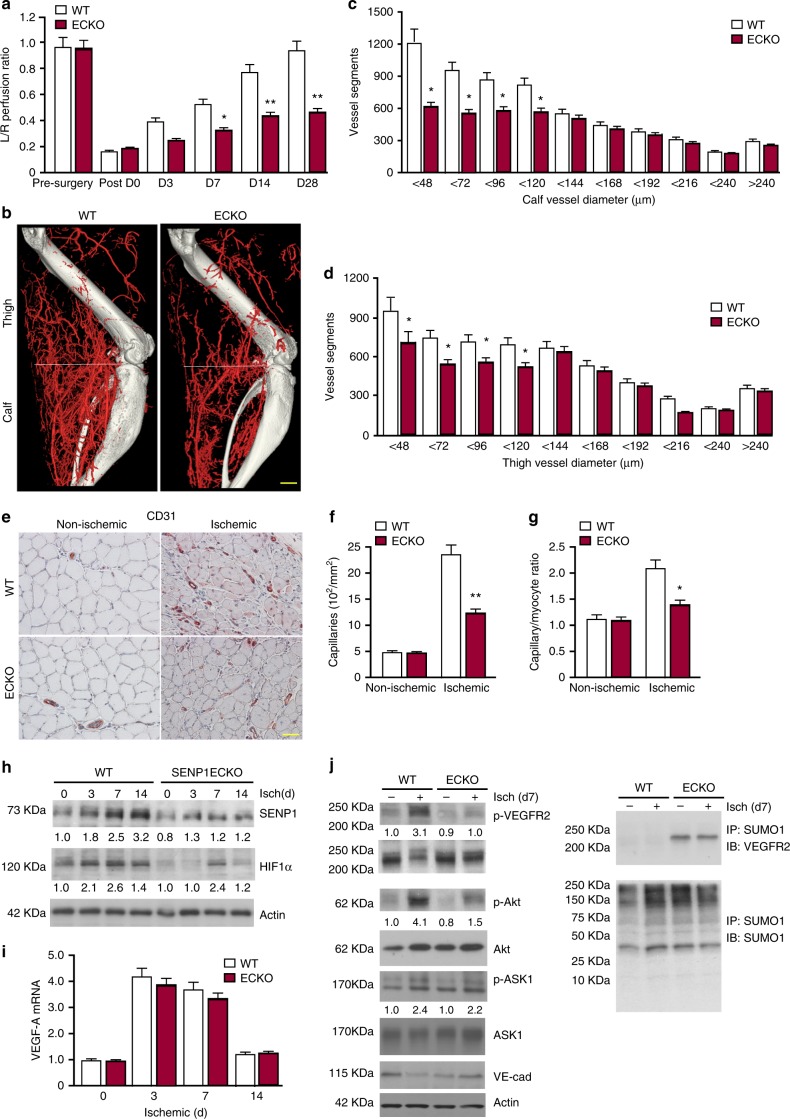
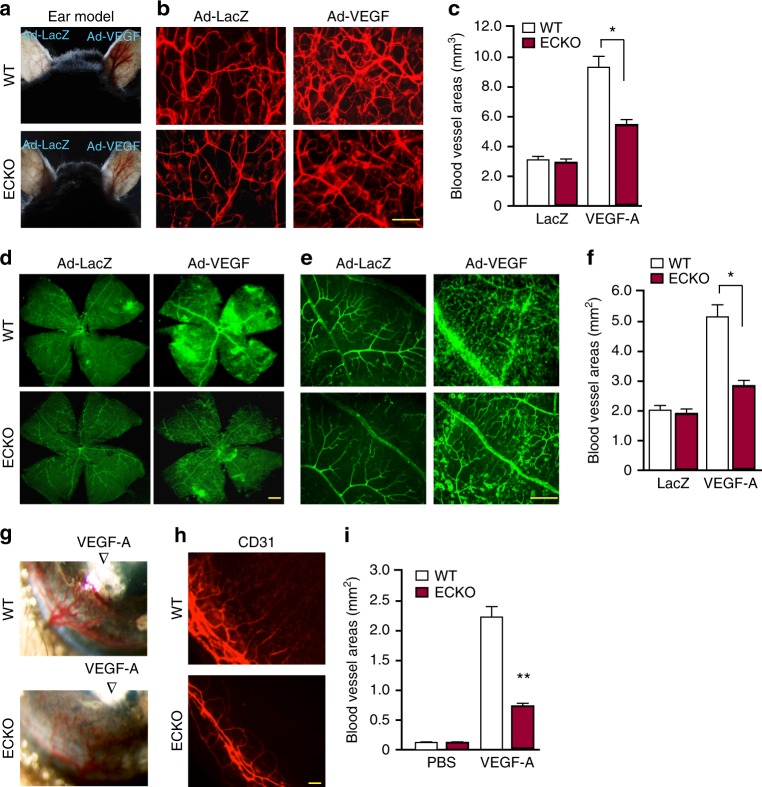
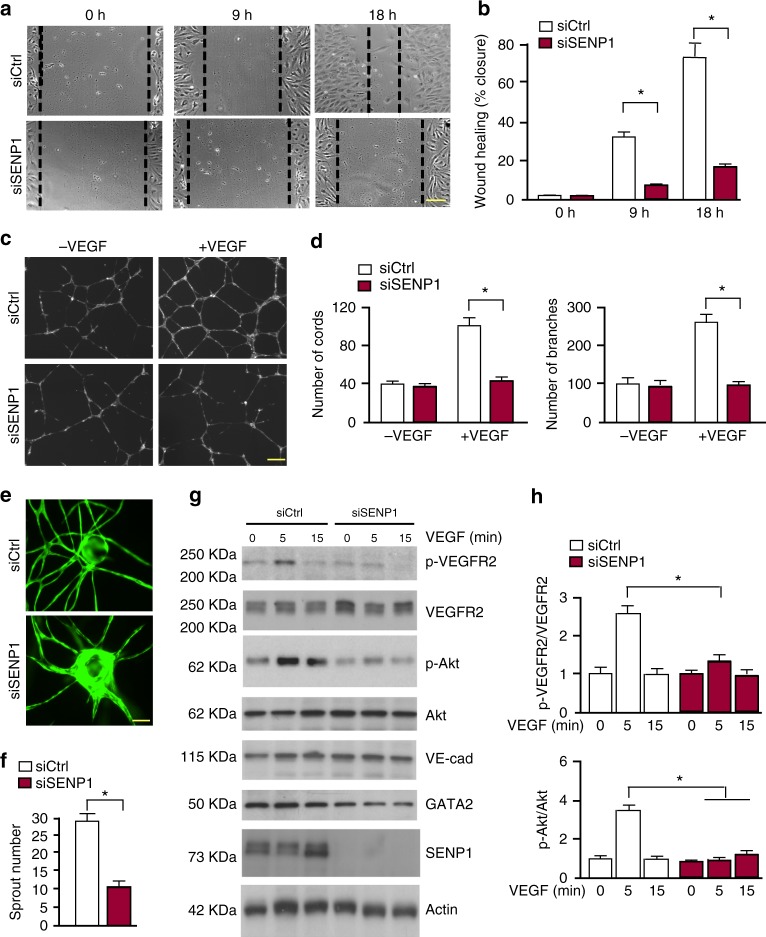
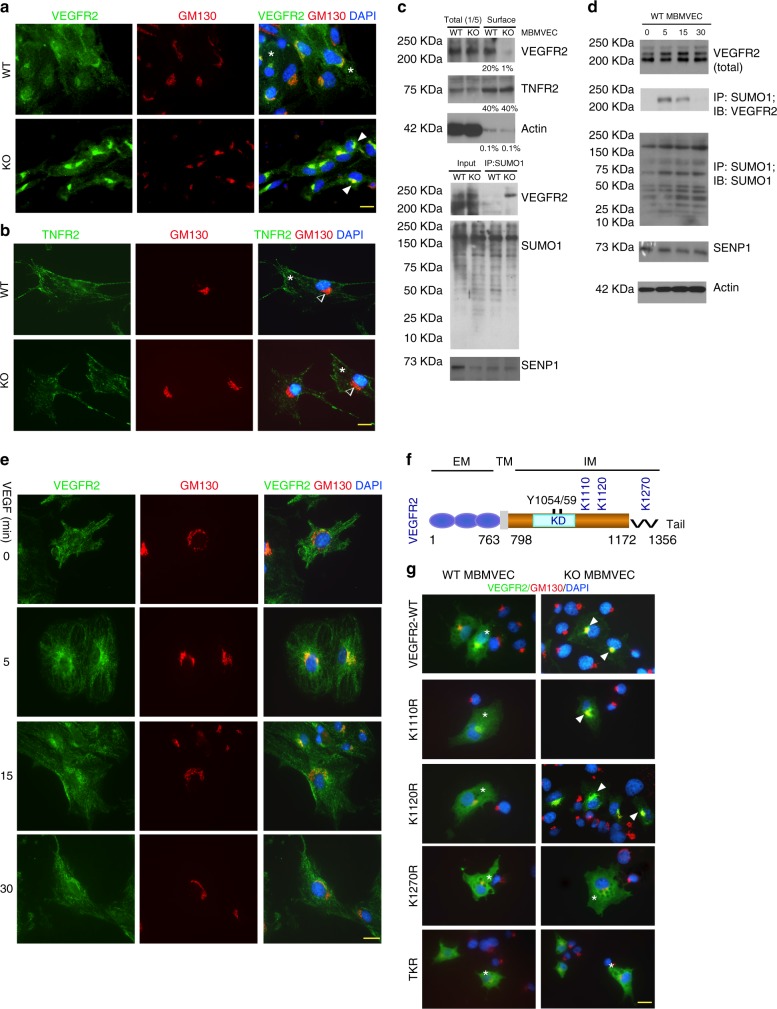
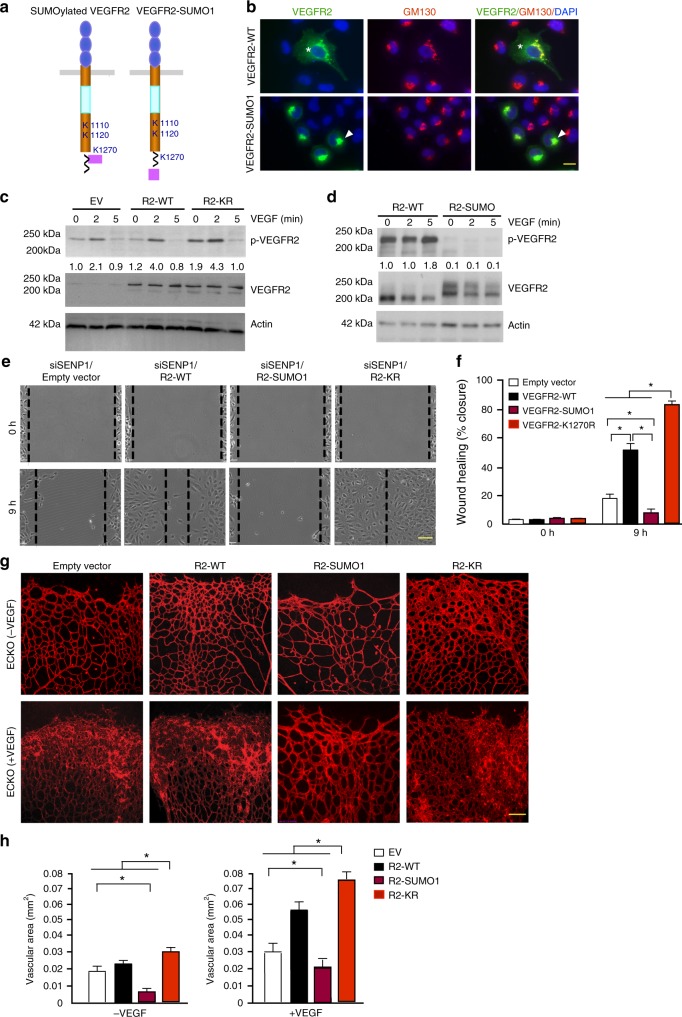
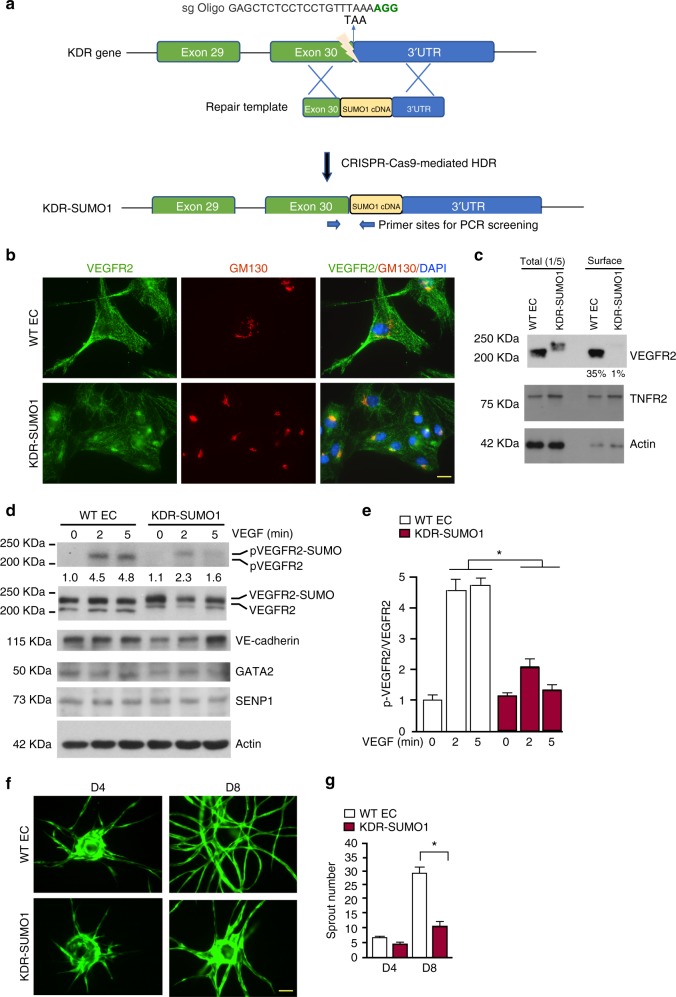
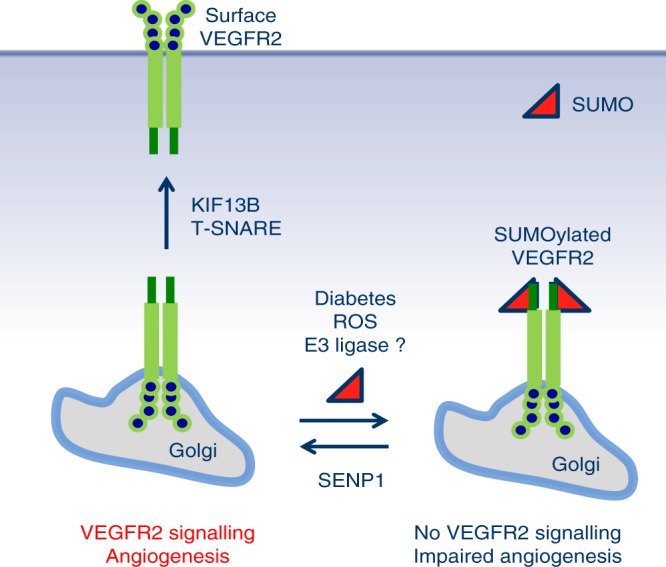
Regulation of VEGFR2 represents an important mechanism for the control of angiogenesis. VEGFR2 activity can be regulated by post-translational modifications such as ubiquitination and acetylation. However, whether VEGFR2 can be regulated by SUMOylation has not been investigated. Here we show that endothelial-specific deletion of the SUMO endopeptidase SENP1 reduces pathological angiogenesis and tissue repair during hindlimb ischemia, and VEGF-induced angiogenesis in the cornea, retina, and ear. SENP1-deficient endothelial cells show increased SUMOylation of VEGFR2 and impaired VEGFR2 signalling. SUMOylation at lysine 1270 retains VEGFR2 in the Golgi and reduces its surface expression, attenuating VEGFR2-dependent signalling. Moreover, we find that SENP1 is downregulated and VEGFR2 hyper-SUMOylated in diabetic settings and that expression of a non-SUMOylated form of VEGFR2 rescues angiogenic defects in diabetic mice. These results show that VEGFR2 is regulated by deSUMOylation during pathological angiogenesis, and propose SENP1 as a potential therapeutic target for the treatment of diabetes-associated angiogenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL136507/HL/NHLBI NIH HHS/United States
- R01 HL109420/HL/NHLBI NIH HHS/United States
- HL109420/U.S. Department of Health & Human Services | NIH | Center for Scientific Review (CSR)/International
- R01 HL115148/HL/NHLBI NIH HHS/United States
- HL115148/U.S. Department of Health & Human Services | NIH | Center for Scientific Review (CSR)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
