

Uridylation by TUT4/7 Restricts Retrotransposition of Human LINE-1s
- PMID: 30122351
- PMCID: PMC6191937
- DOI: 10.1016/j.cell.2018.07.022
Uridylation by TUT4/7 Restricts Retrotransposition of Human LINE-1s
Abstract
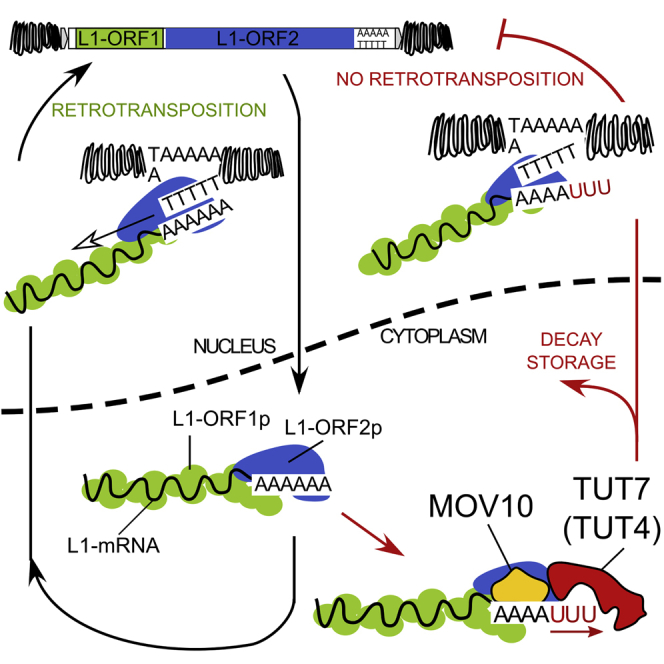
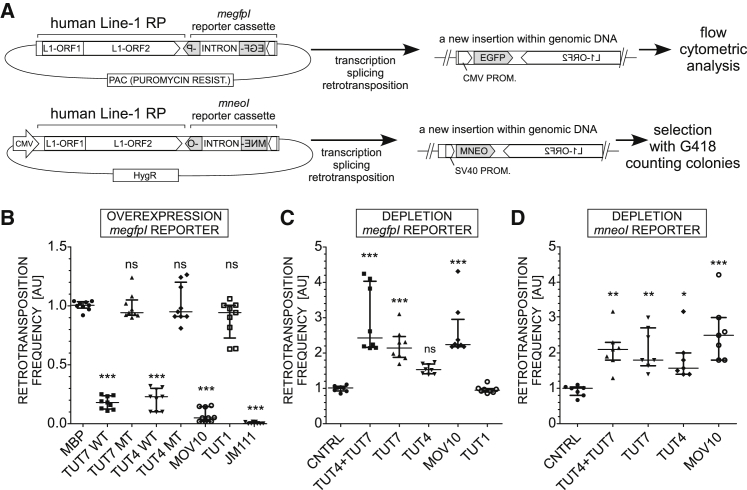
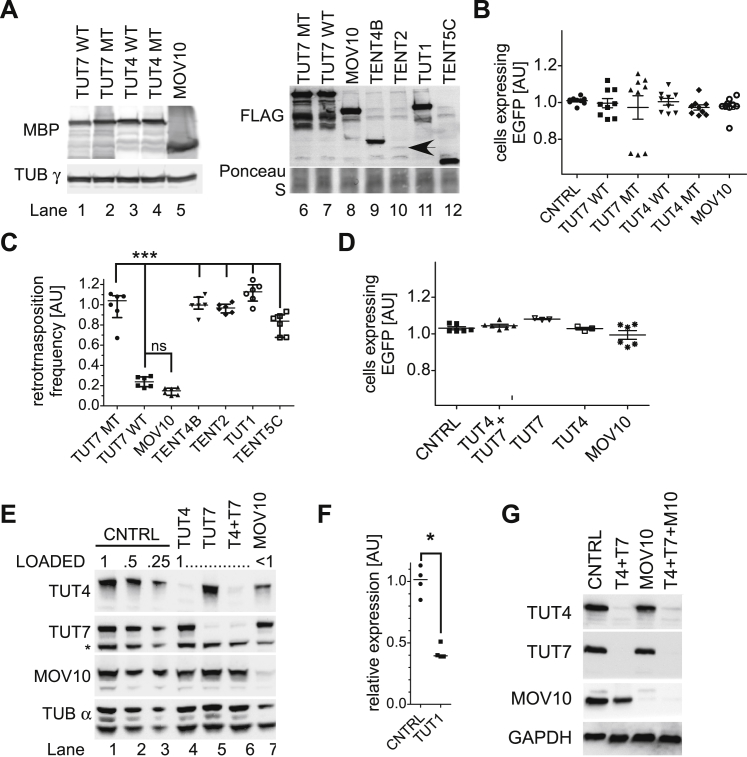
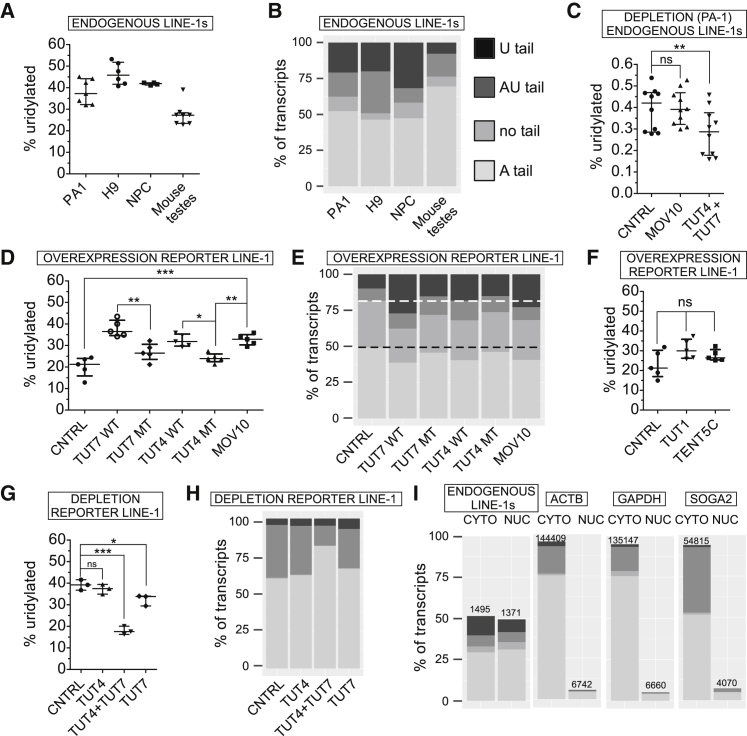
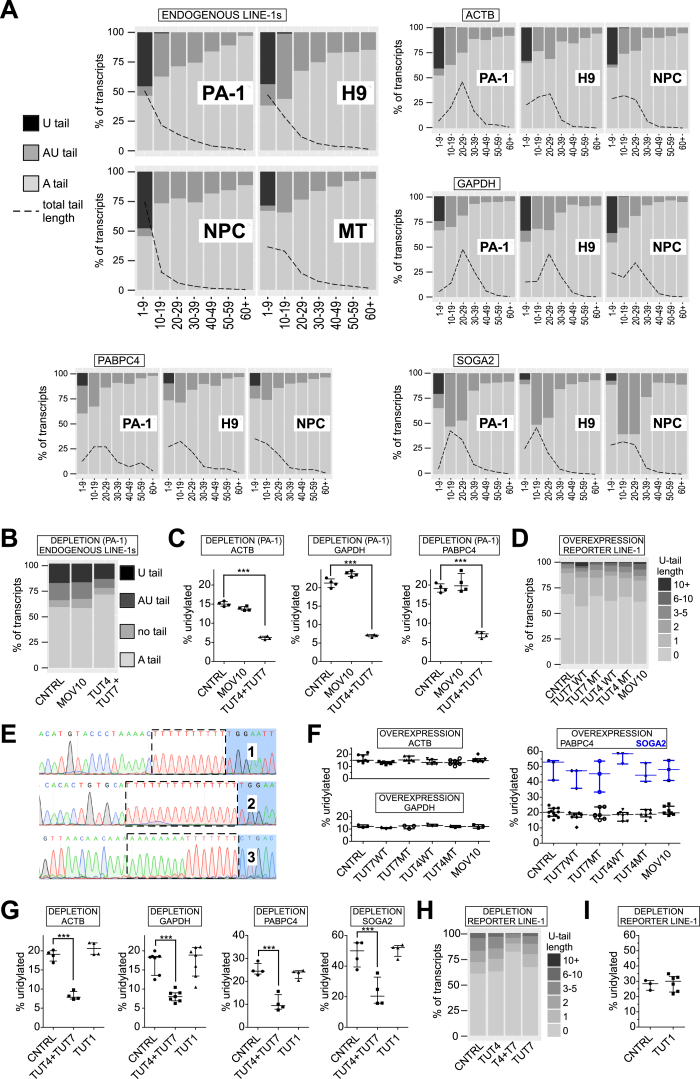
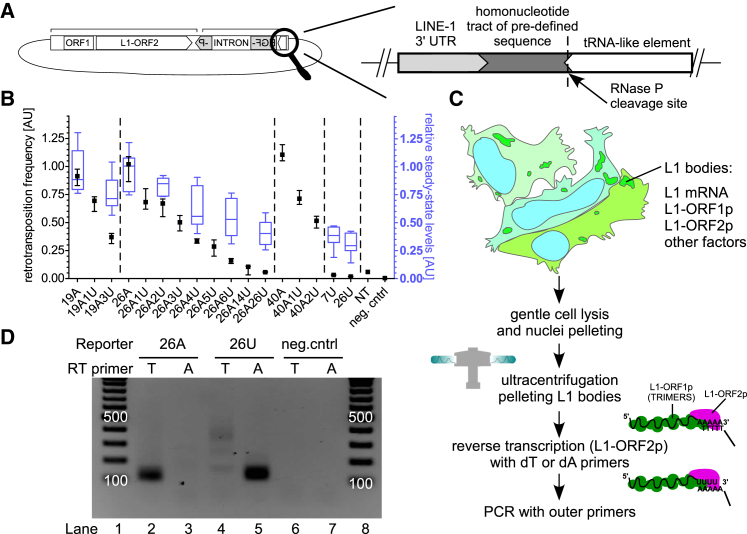
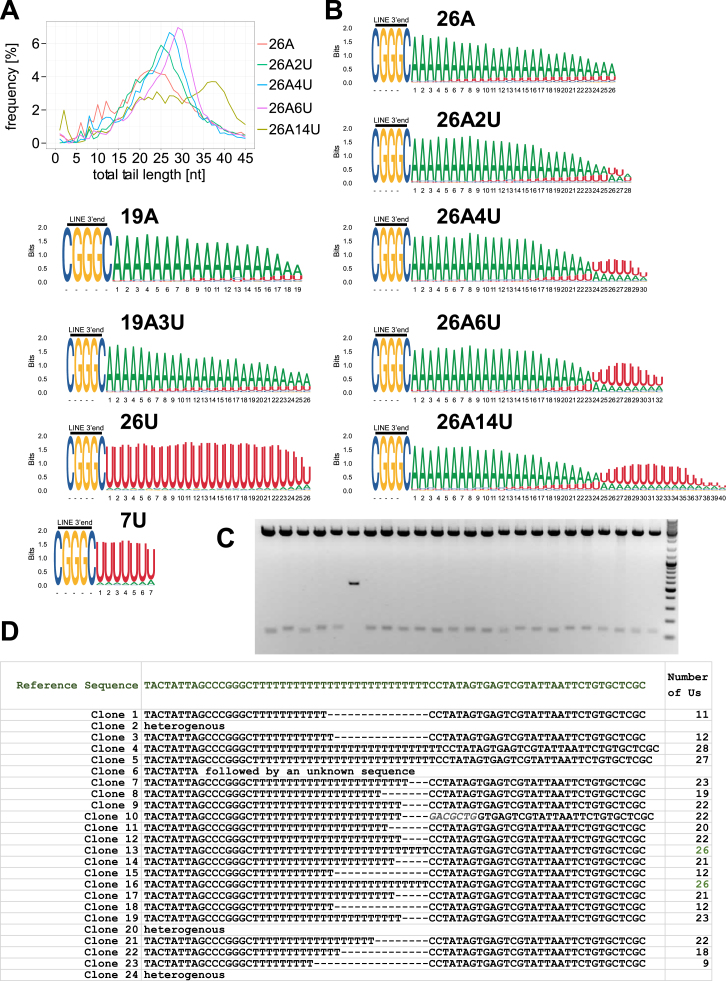
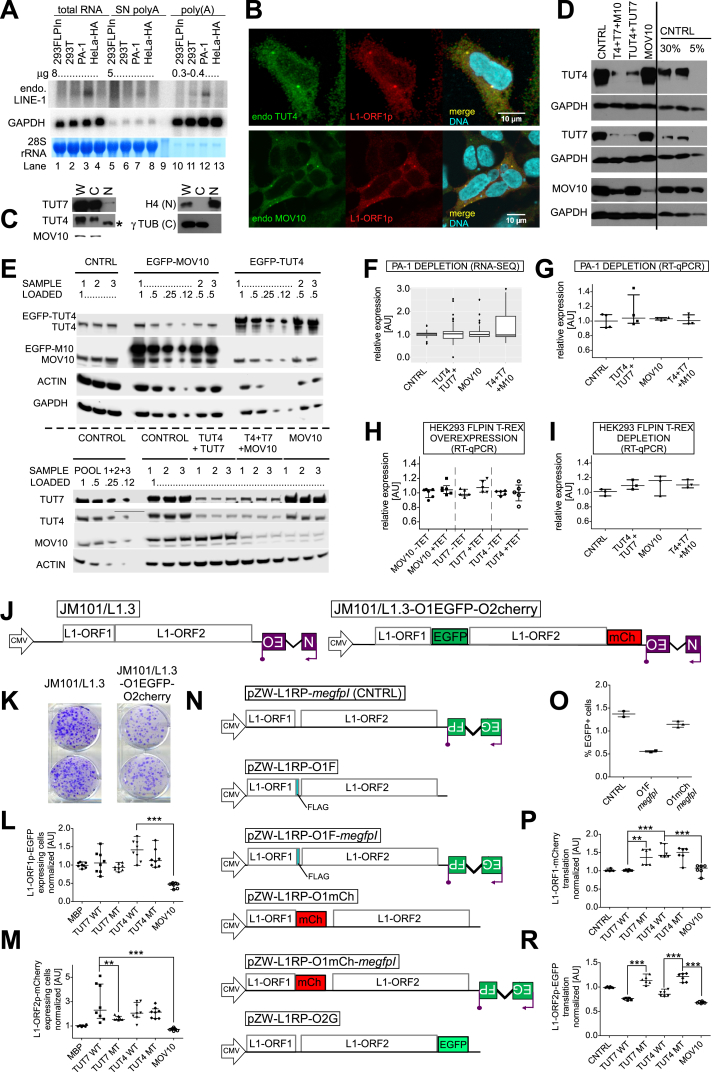
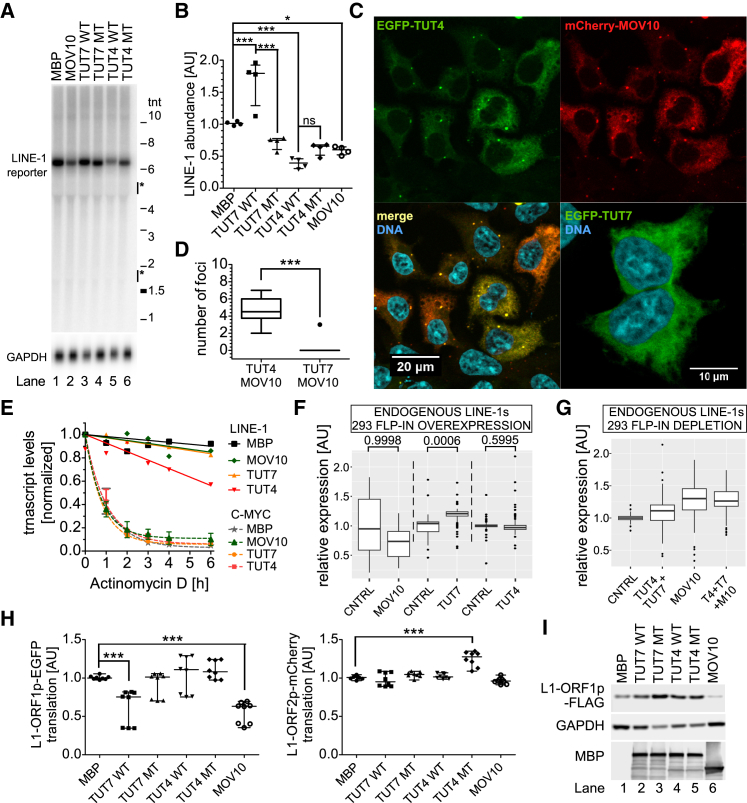
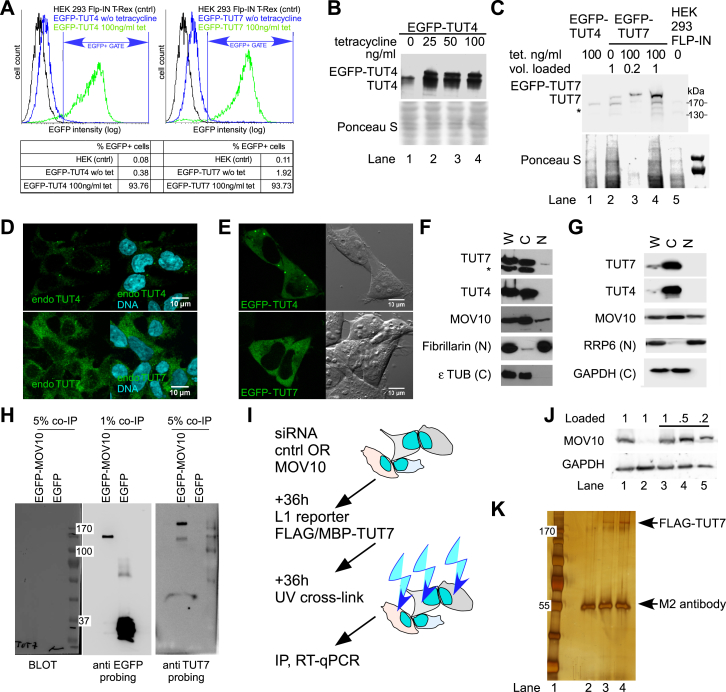
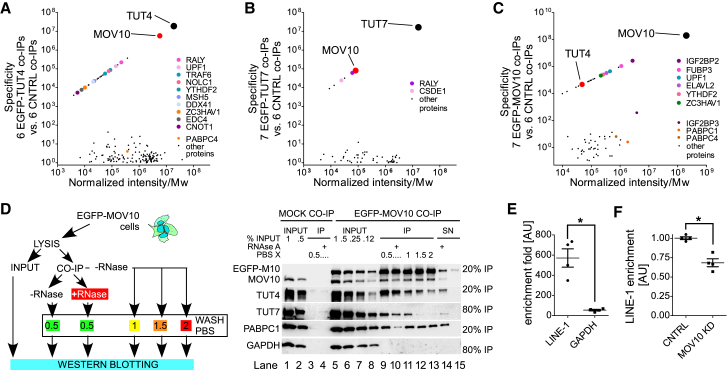
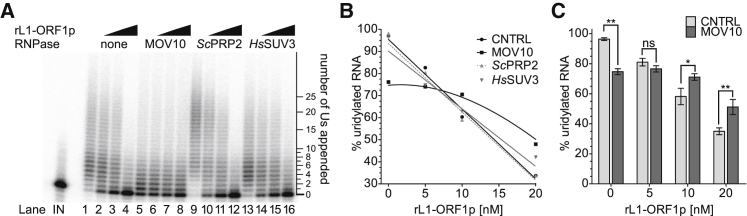
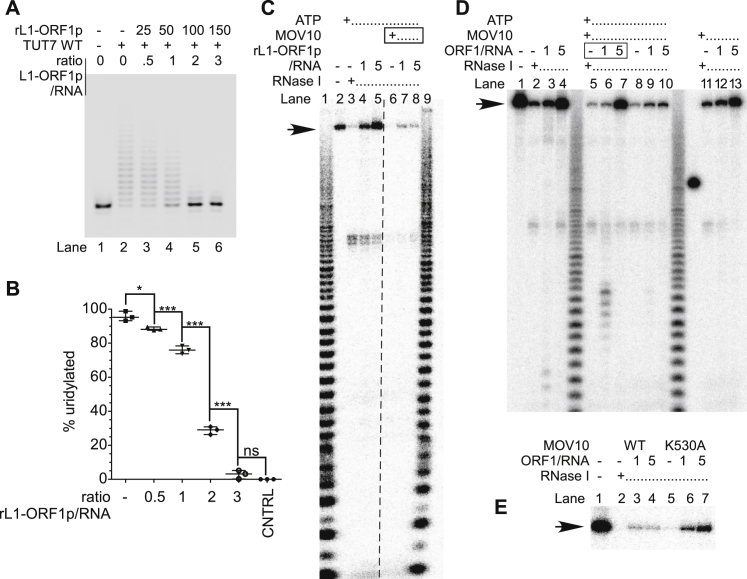
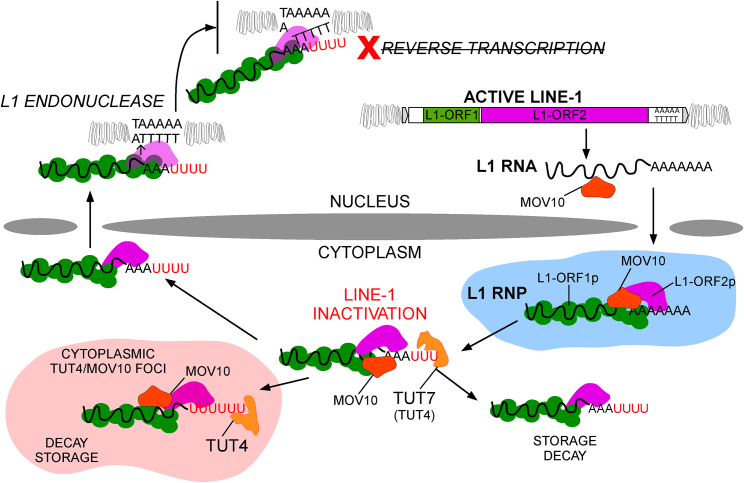
LINE-1 retrotransposition is tightly restricted by layers of regulatory control, with epigenetic pathways being the best characterized. Looking at post-transcriptional regulation, we now show that LINE-1 mRNA 3' ends are pervasively uridylated in various human cellular models and in mouse testes. TUT4 and TUT7 uridyltransferases catalyze the modification and function in cooperation with the helicase/RNPase MOV10 to counteract the RNA chaperone activity of the L1-ORF1p retrotransposon protein. Uridylation potently restricts LINE-1 retrotransposition by a multilayer mechanism depending on differential subcellular localization of the uridyltransferases. We propose that uridine residues added by TUT7 in the cytoplasm inhibit initiation of reverse transcription of LINE-1 mRNAs once they are reimported to the nucleus, whereas uridylation by TUT4, which is enriched in cytoplasmic foci, destabilizes mRNAs. These results provide a model for the post-transcriptional restriction of LINE-1, revealing a key physiological role for TUT4/7-mediated uridylation in maintaining genome stability.
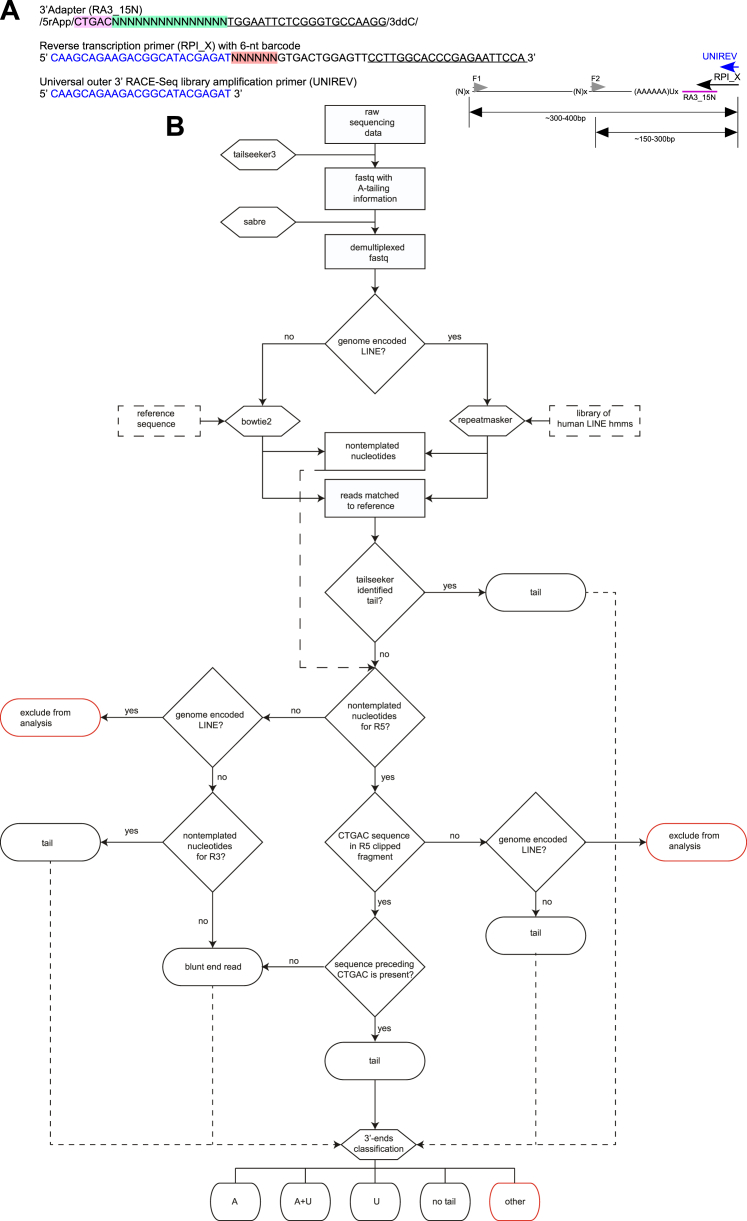
Keywords: 3′ RACE-seq; L1-ORF1p; L1-ORF2p; LINE-1; MOV10; TUT4; TUT7; poly(A); retrotransposition; uridylation.
Copyright © 2018 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
TUT-TUTting retrotransposons.Nat Rev Mol Cell Biol. 2018 Oct;19(10):618. doi: 10.1038/s41580-018-0058-2. Nat Rev Mol Cell Biol. 2018. PMID: 30158668 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
