

Long QT Syndrome and Sinus Bradycardia-A Mini Review
- PMID: 30123799
- PMCID: PMC6085426
- DOI: 10.3389/fcvm.2018.00106
Long QT Syndrome and Sinus Bradycardia-A Mini Review
Abstract
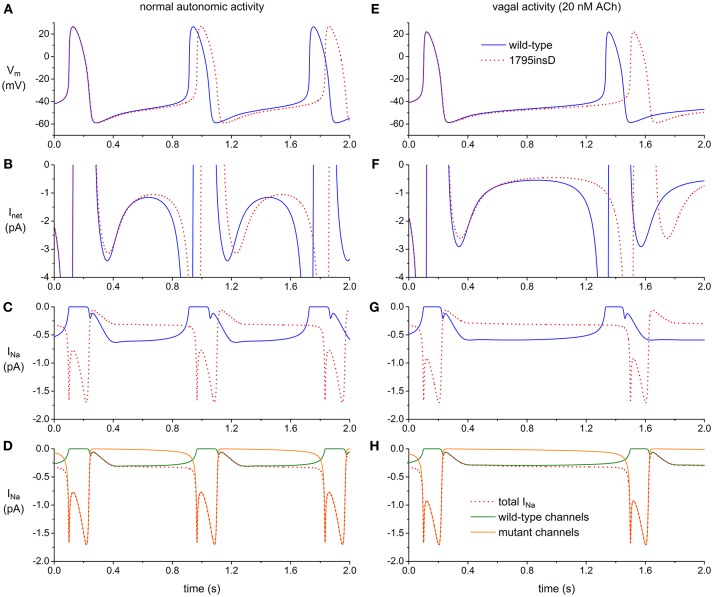
Congenital long-QT syndrome (LQTS) is an inherited cardiac disorder characterized by the prolongation of ventricular repolarization, susceptibility to Torsades de Pointes (TdP), and a risk for sudden death. Various types of congenital LQTS exist, all due to specific defects in ion channel-related genes. Interestingly, almost all of the ion channels affected by the various types of LQTS gene mutations are also expressed in the human sinoatrial node (SAN). It is therefore not surprising that LQTS is frequently associated with a change in basal heart rate (HR). However, current data on how the LQTS-associated ion channel defects result in impaired human SAN pacemaker activity are limited. In this mini-review, we provide an overview of known LQTS mutations with effects on HR and the underlying changes in expression and kinetics of ion channels. Sinus bradycardia has been reported in relation to a large number of LQTS mutations. However, the occurrence of both QT prolongation and sinus bradycardia on a family basis is almost completely limited to LQTS types 3 and 4 (LQT3 and Ankyrin-B syndrome, respectively). Furthermore, a clear causative role of this sinus bradycardia in cardiac events seems reserved to mutations underlying LQT3.
Keywords: computer simulation; heart rate; human; ion channel; long-QT syndrome; mutations; sinoatrial node; sinus bradycardia.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources