

Hypermorphic and hypomorphic AARS alleles in patients with CMT2N expand clinical and molecular heterogeneities
- PMID: 30124830
- PMCID: PMC6240730
- DOI: 10.1093/hmg/ddy290
Hypermorphic and hypomorphic AARS alleles in patients with CMT2N expand clinical and molecular heterogeneities
Abstract
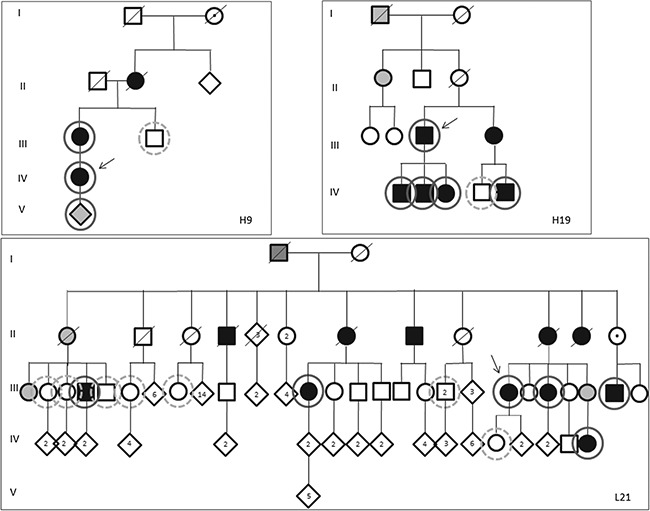
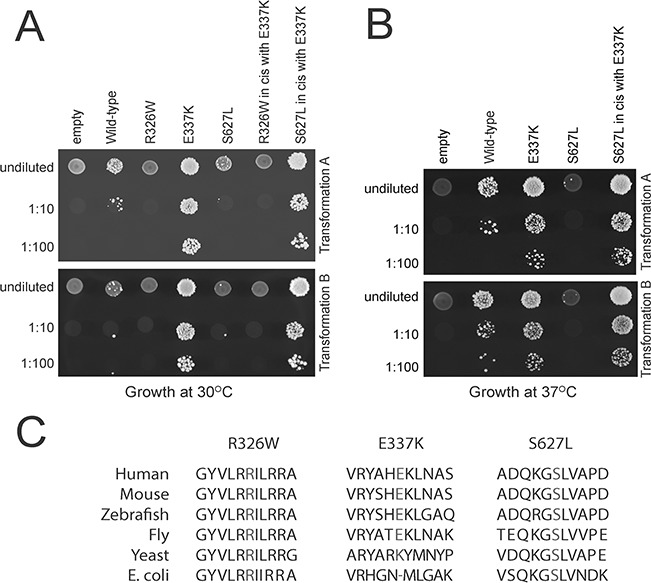
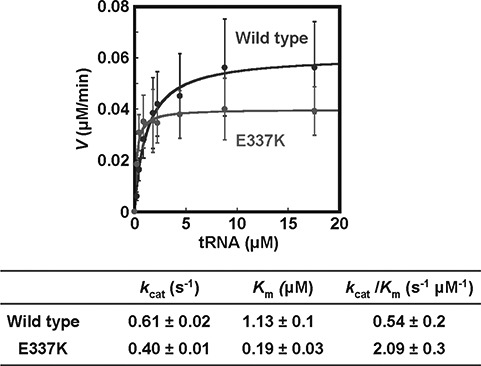
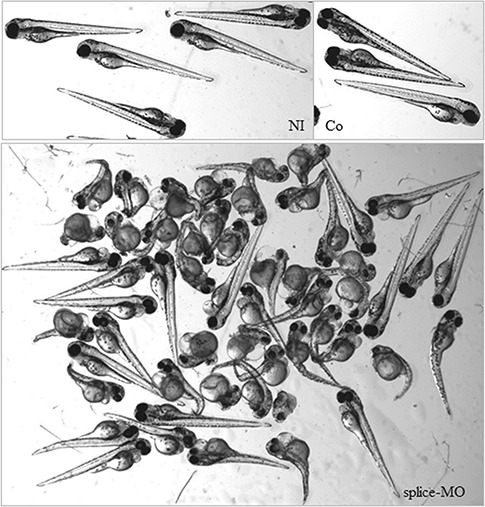
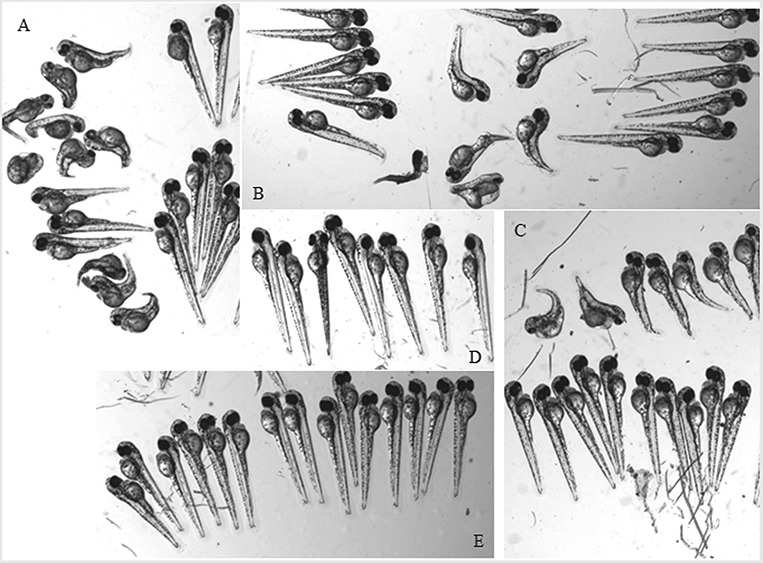
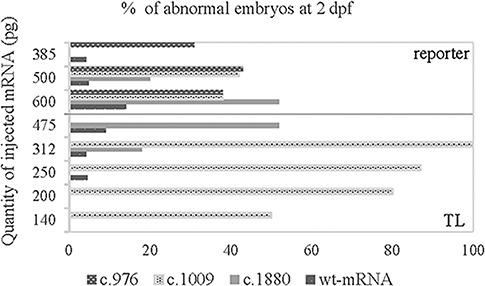
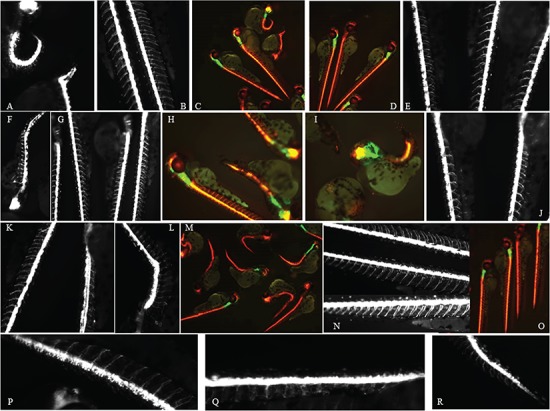
Aminoacyl-tRNA synthetases (ARSs) are ubiquitously expressed enzymes implicated in several dominant and recessive disease phenotypes. The canonical function of ARSs is to couple an amino acid to a cognate transfer RNA (tRNA). We identified three novel disease-associated missense mutations in the alanyl-tRNA synthetase (AARS) gene in three families with dominant axonal Charcot-Marie-Tooth (CMT) disease. Two mutations (p.Arg326Trp and p.Glu337Lys) are located near a recurrent pathologic change in AARS, p.Arg329His. The third (p.Ser627Leu) is in the editing domain of the protein in which hitherto only mutations associated with recessive encephalopathies have been described. Yeast complementation assays demonstrated that two mutations (p.Ser627Leu and p.Arg326Trp) represent loss-of-function alleles, while the third (p.Glu337Lys) represents a hypermorphic allele. Further, aminoacylation assays confirmed that the third mutation (p.Glu337Lys) increases tRNA charging velocity. To test the effect of each mutation in the context of a vertebrate nervous system, we developed a zebrafish assay. Remarkably, all three mutations caused a pathological phenotype of neural abnormalities when expressed in zebrafish, while expression of the human wild-type messenger RNA (mRNA) did not. Our data indicate that not only functional null or hypomorphic alleles, but also hypermorphic AARS alleles can cause dominantly inherited axonal CMT disease.
Figures
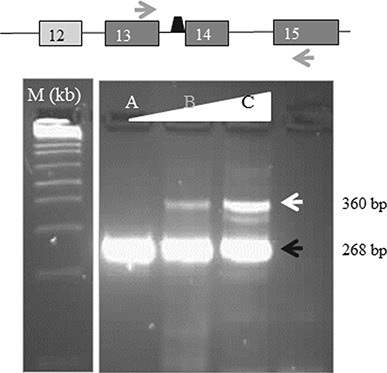
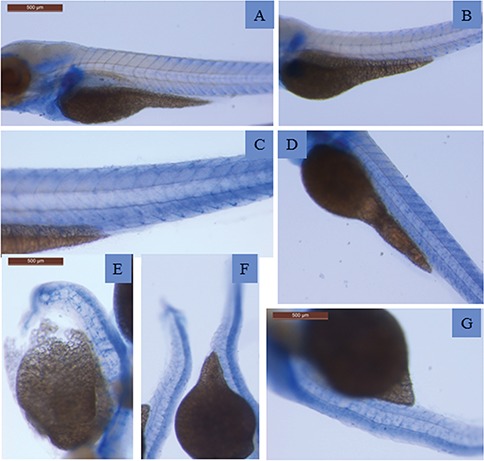
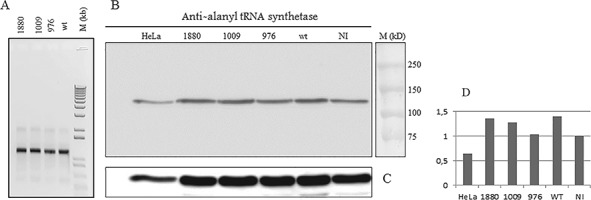
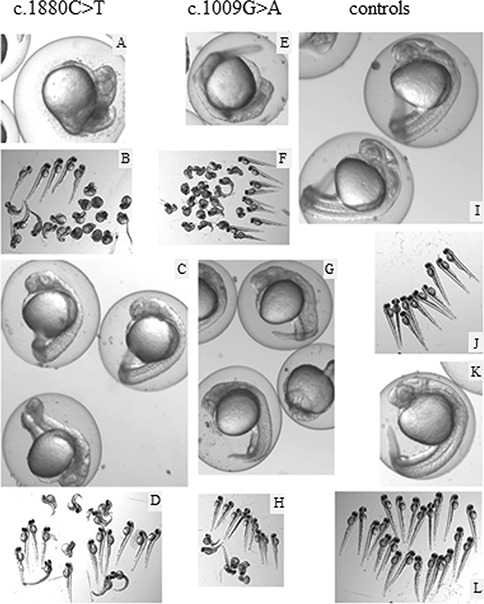
References
-
- Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. et al. (2003) Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet., 72, 1293–1299. - PMC - PubMed
-
- Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V. et al. (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet., 38, 197–202. - PubMed
-
- Latour P., Thauvin-Robinet C., Baudelet-Mery C., Soichot P., Cusin V., Faivre L., Locatelli M.C., Mayencon M., Sarcey A., Broussolle E. et al. (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet., 86, 77–82. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
