

Comparative genomic analysis of Staphylococcus lugdunensis shows a closed pan-genome and multiple barriers to horizontal gene transfer
- PMID: 30126366
- PMCID: PMC6102843
- DOI: 10.1186/s12864-018-4978-1
Comparative genomic analysis of Staphylococcus lugdunensis shows a closed pan-genome and multiple barriers to horizontal gene transfer
Abstract
Background: Coagulase negative staphylococci (CoNS) are commensal bacteria on human skin. Staphylococcus lugdunensis is a unique CoNS which produces various virulence factors and may, like S. aureus, cause severe infections, particularly in hospital settings. Unlike other staphylococci, it remains highly susceptible to antimicrobials, and genome-based phylogenetic studies have evidenced a highly conserved genome that distinguishes it from all other staphylococci.
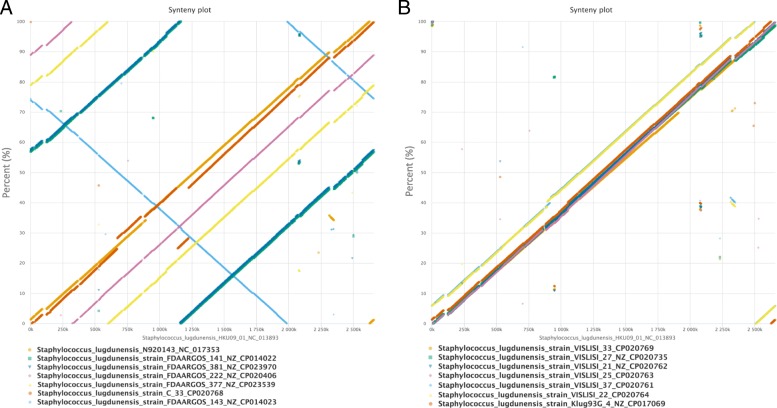
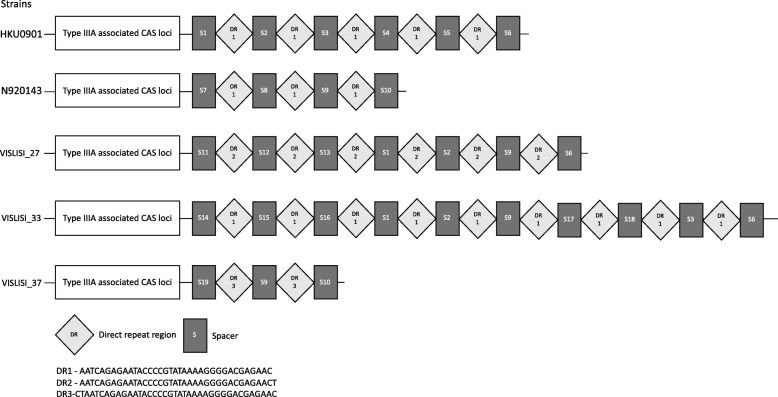
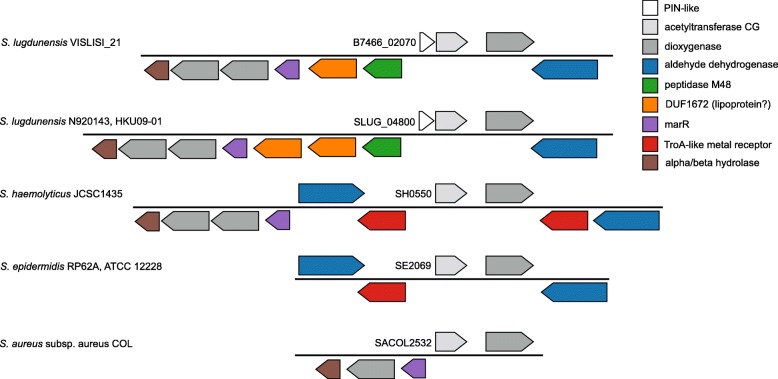
Results: We demonstrate that S. lugdunensis possesses a closed pan-genome with a very limited number of new genes, in contrast to other staphylococci that have an open pan-genome. Whole-genome nucleotide and amino acid identity levels are also higher than in other staphylococci. We identified numerous genetic barriers to horizontal gene transfer that might explain this result. The S. lugdunensis genome has multiple operons encoding for restriction-modification, CRISPR/Cas and toxin/antitoxin systems. We also identified a new PIN-like domain-associated protein that might belong to a larger operon, comprising a metalloprotease, that could function as a new toxin/antitoxin or detoxification system.
Conclusion: We show that S. lugdunensis has a unique genome profile within staphylococci, with a closed pan-genome and several systems to prevent horizontal gene transfer. Its virulence in clinical settings does not rely on its ability to acquire and exchange antibiotic resistance genes or other virulence factors as shown for other staphylococci.
Keywords: CRISPR; Comparative genomics; Core genome; Pan genome; Restriction-modification; Staphylococcus lugdunensis; Toxin/antitoxin.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



References
-
- Argemi X, Riegel P, Lavigne T, Lefebvre N, Grandpré N, Hansmann Y, et al. Implementation of MALDI-TOF MS in routine clinical laboratories improves identification of coagulase negative staphylococci and reveals the pathogenic role of Staphylococcus lugdunensis. J Clin Microbiol. 2015;53(7):2030–2036. doi: 10.1128/JCM.00177-15. - DOI - PMC - PubMed
-
- Argemi X, Prévost G, Riegel P, Keller D, Meyer N, Baldeyrou M, et al. VISLISI trial, a prospective clinical study allowing identification of a new metalloprotease and putative virulence factor from Staphylococcus lugdunensis. Clin Microbiol Infect Off Publ Eur Soc Clin Microbiol Infect Dis. 2017;23:334. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
