

Long-read sequencing across the C9orf72 'GGGGCC' repeat expansion: implications for clinical use and genetic discovery efforts in human disease
- PMID: 30126445
- PMCID: PMC6102925
- DOI: 10.1186/s13024-018-0274-4
Long-read sequencing across the C9orf72 'GGGGCC' repeat expansion: implications for clinical use and genetic discovery efforts in human disease
Abstract
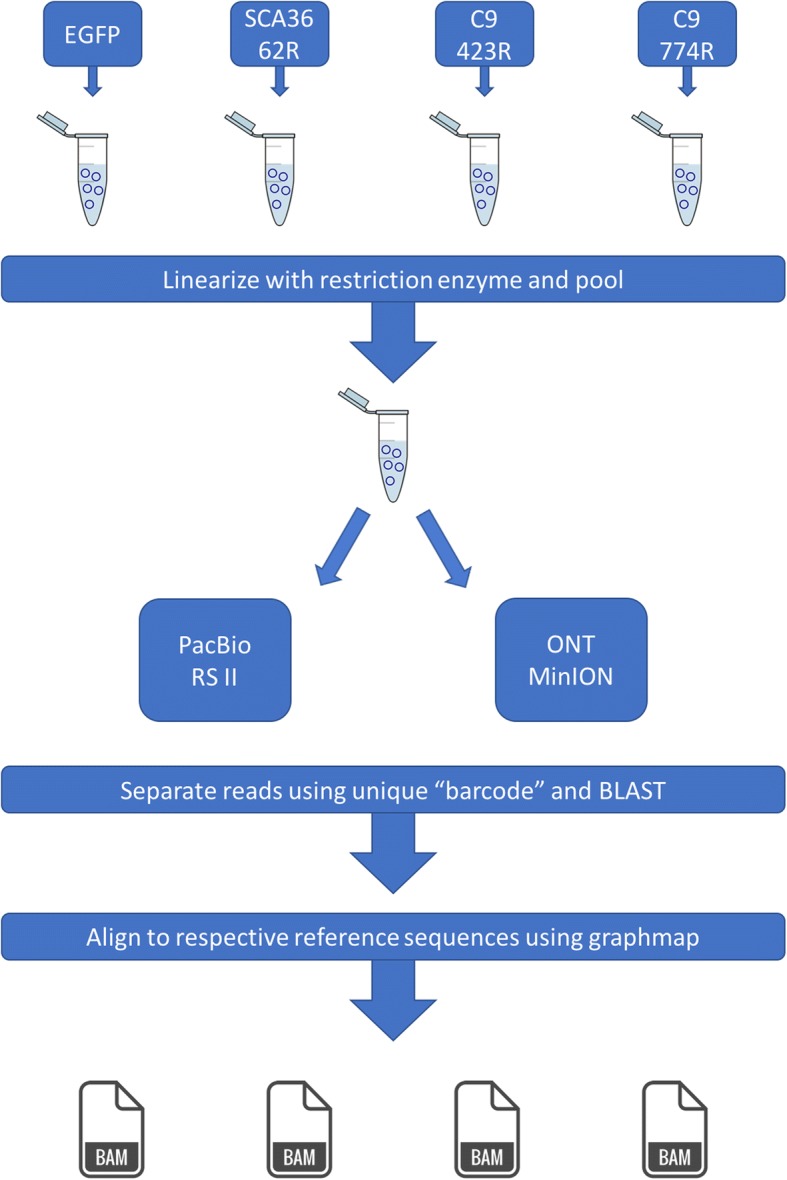
Background: Many neurodegenerative diseases are caused by nucleotide repeat expansions, but most expansions, like the C9orf72 'GGGGCC' (G4C2) repeat that causes approximately 5-7% of all amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) cases, are too long to sequence using short-read sequencing technologies. It is unclear whether long-read sequencing technologies can traverse these long, challenging repeat expansions. Here, we demonstrate that two long-read sequencing technologies, Pacific Biosciences' (PacBio) and Oxford Nanopore Technologies' (ONT), can sequence through disease-causing repeats cloned into plasmids, including the FTD/ALS-causing G4C2 repeat expansion. We also report the first long-read sequencing data characterizing the C9orf72 G4C2 repeat expansion at the nucleotide level in two symptomatic expansion carriers using PacBio whole-genome sequencing and a no-amplification (No-Amp) targeted approach based on CRISPR/Cas9.
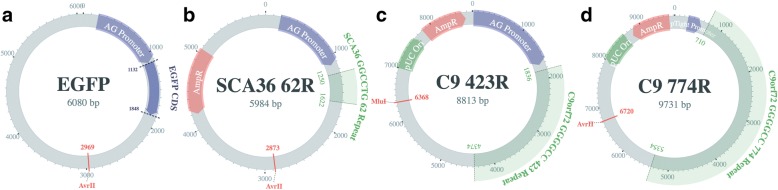
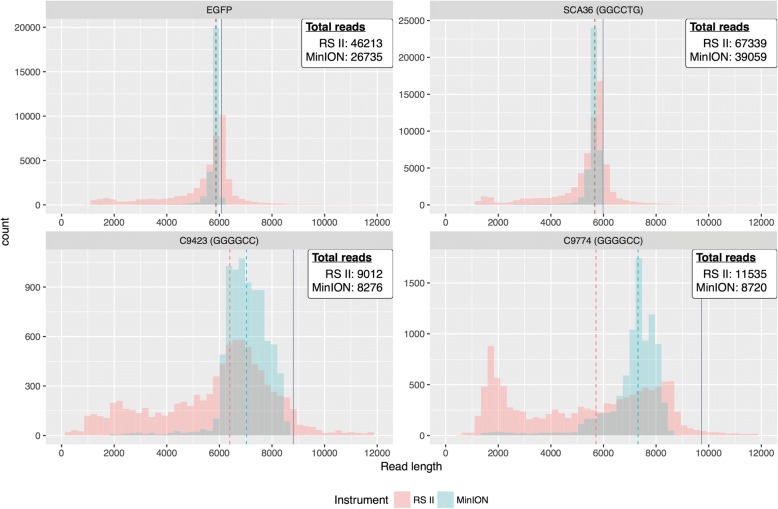
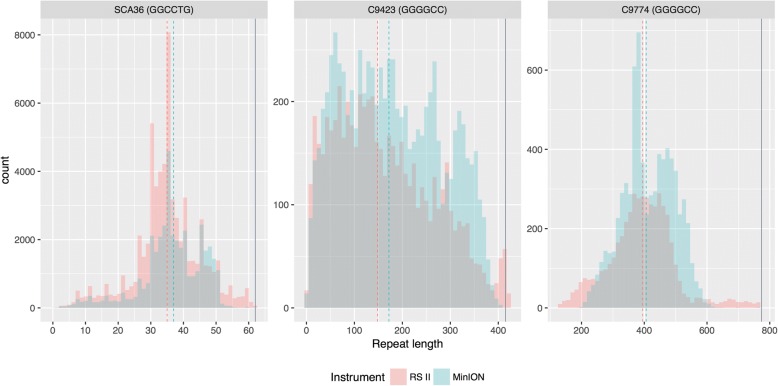
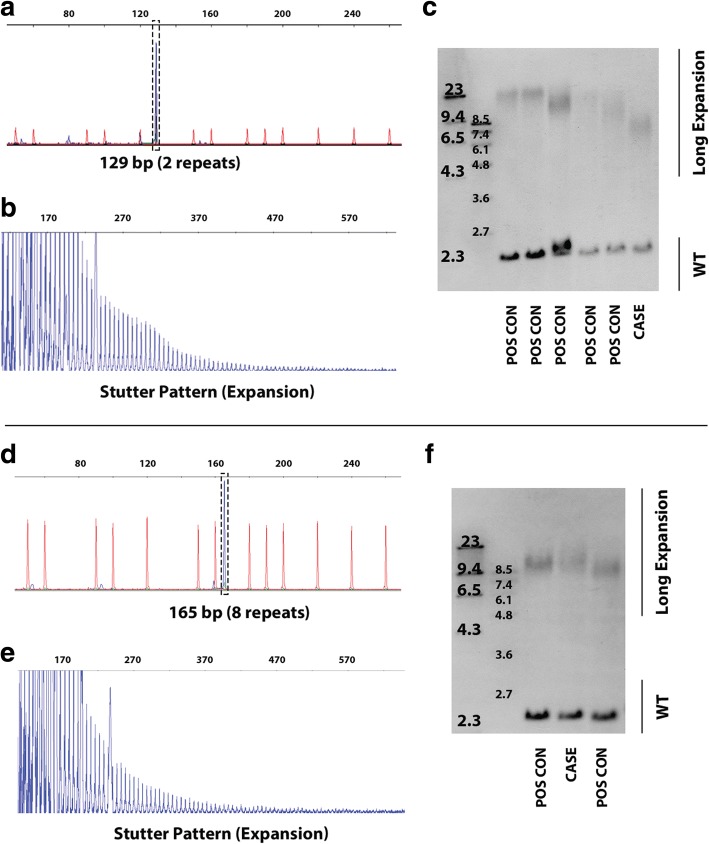
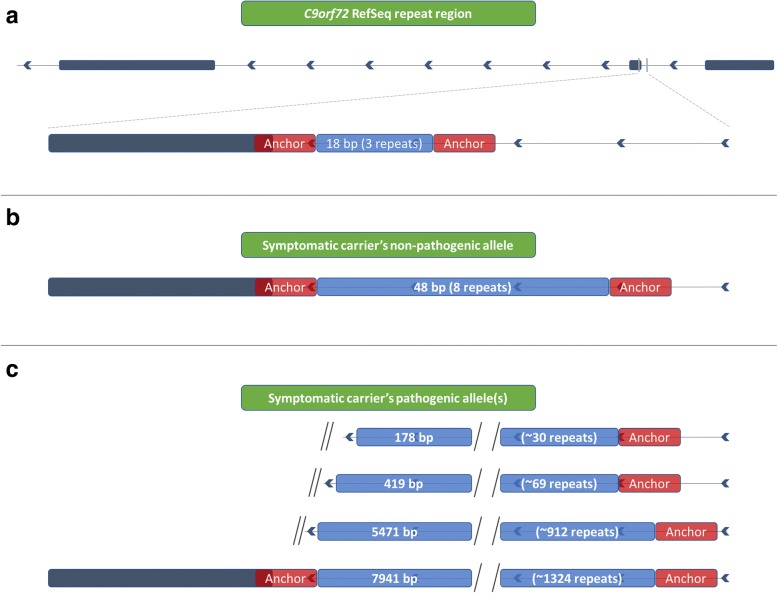
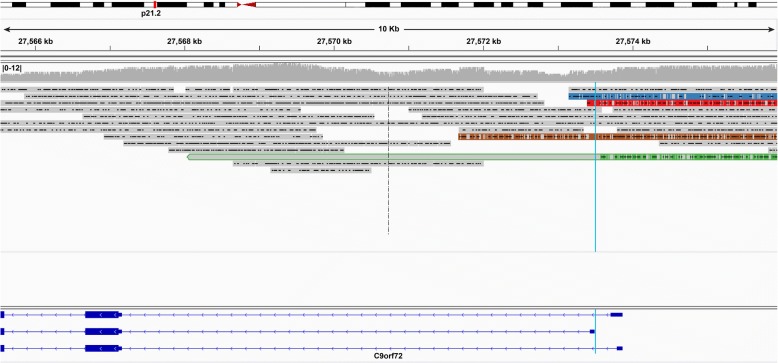
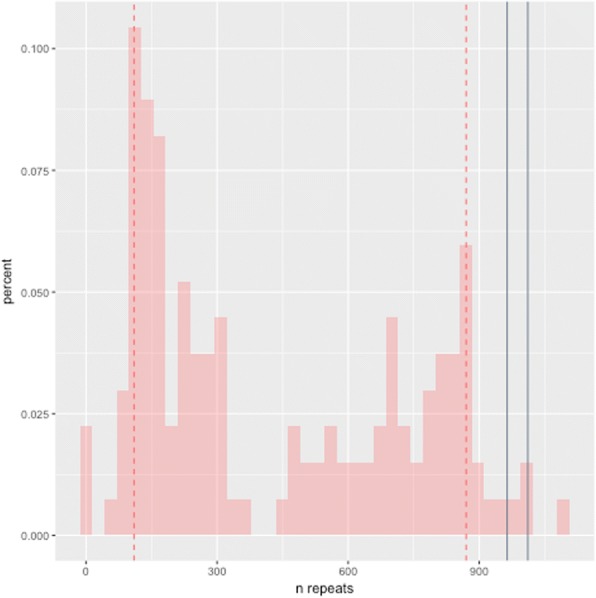
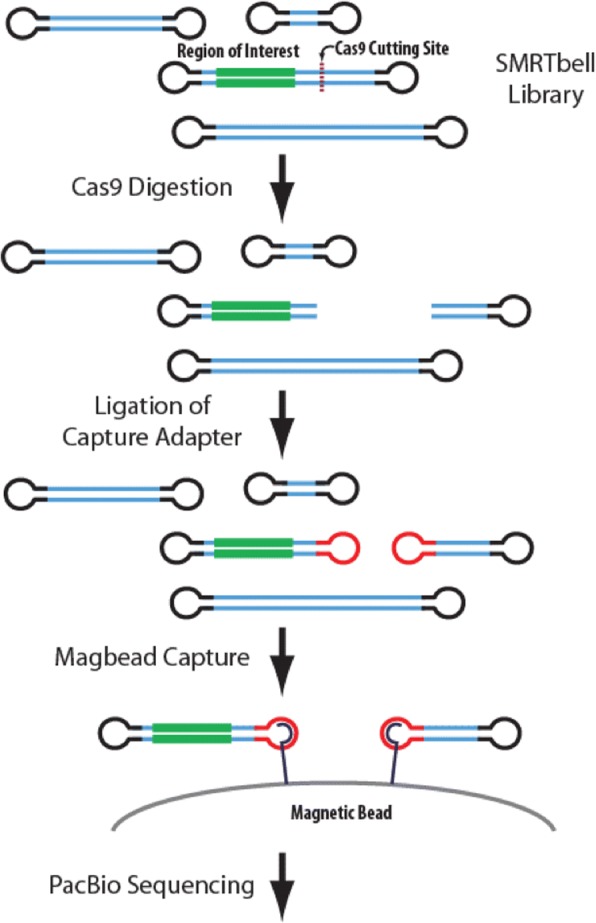
Results: Both the PacBio and ONT platforms successfully sequenced through the repeat expansions in plasmids. Throughput on the MinION was a challenge for whole-genome sequencing; we were unable to attain reads covering the human C9orf72 repeat expansion using 15 flow cells. We obtained 8× coverage across the C9orf72 locus using the PacBio Sequel, accurately reporting the unexpanded allele at eight repeats, and reading through the entire expansion with 1324 repeats (7941 nucleotides). Using the No-Amp targeted approach, we attained > 800× coverage and were able to identify the unexpanded allele, closely estimate expansion size, and assess nucleotide content in a single experiment. We estimate the individual's repeat region was > 99% G4C2 content, though we cannot rule out small interruptions.
Conclusions: Our findings indicate that long-read sequencing is well suited to characterizing known repeat expansions, and for discovering new disease-causing, disease-modifying, or risk-modifying repeat expansions that have gone undetected with conventional short-read sequencing. The PacBio No-Amp targeted approach may have future potential in clinical and genetic counseling environments. Larger and deeper long-read sequencing studies in C9orf72 expansion carriers will be important to determine heterogeneity and whether the repeats are interrupted by non-G4C2 content, potentially mitigating or modifying disease course or age of onset, as interruptions are known to do in other repeat-expansion disorders. These results have broad implications across all diseases where the genetic etiology remains unclear.
Keywords: Amyotrophic lateral sclerosis (ALS); C9orf72; Frontotemporal dementia (FTD); GGGGCC; Genetics; Long-read sequencing; Oxford Nanopore Technologies MinION; PacBio RS II and Sequel; Repeat expansion disorders; Structural mutations.
Conflict of interest statement
Ethics approval and consent to participate
The Mayo Clinic Institutional Review Board (IRB) approved all procedures for this study and we followed all appropriate protocols.
Consent for publication
All participants were properly consented for this study.
Competing interests
IM, BB, and MS are full-time employees of Pacific Biosciences of California, Inc. IM’s spouse is also a full-time employee of, and owns stock in, Pacific Biosciences of California, Inc. All other authors declare they have no conflicts of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in non-coding region of C9ORF72 causes chromosome 9p-linked frontotemporal dementia and amyotrophic lateral sclerosis. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R35 NS097273/NS/NINDS NIH HHS/United States
- R21 AG047327/AG/NIA NIH HHS/United States
- NS097261/NS/NINDS NIH HHS/United States
- NS099631/NS/NINDS NIH HHS/United States
- AG047327/AG/NIA NIH HHS/United States
- NS084974/NS/NINDS NIH HHS/United States
- AG049992/AG/NIA NIH HHS/United States
- NS094137/NS/NINDS NIH HHS/United States
- R35 NS097261/NS/NINDS NIH HHS/United States
- NS093865/NS/NINDS NIH HHS/United States
- R21 NS099631/NS/NINDS NIH HHS/United States
- 8AZ10/Florida Department of Health/International
- R01 NS093865/NS/NINDS NIH HHS/United States
- 6AZ06/Florida Department of Health/International
- R03 AG049992/AG/NIA NIH HHS/United States
- NS097273/NS/NINDS NIH HHS/United States
- R01 NS088689/NS/NINDS NIH HHS/United States
- AL130125/U.S. Department of Defense/International
- P01 NS084974/NS/NINDS NIH HHS/United States
- R21 NS084528/NS/NINDS NIH HHS/United States
- NS088689/NS/NINDS NIH HHS/United States
- NS099114/NS/NINDS NIH HHS/United States
- P01 NS099114/NS/NINDS NIH HHS/United States
- NS084528/National Institute of Neurological Disorders and Stroke (US)/International
- R01 NS094137/NS/NINDS NIH HHS/United States
- RSGTMT17/Pharmaceutical Research and Manufacturers of America Foundation/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
