

Proteome-wide analysis of phospho-regulated PDZ domain interactions
- PMID: 30126976
- PMCID: PMC6100724
- DOI: 10.15252/msb.20178129
Proteome-wide analysis of phospho-regulated PDZ domain interactions
Abstract
A key function of reversible protein phosphorylation is to regulate protein-protein interactions, many of which involve short linear motifs (3-12 amino acids). Motif-based interactions are difficult to capture because of their often low-to-moderate affinities. Here, we describe phosphomimetic proteomic peptide-phage display, a powerful method for simultaneously finding motif-based interaction and pinpointing phosphorylation switches. We computationally designed an oligonucleotide library encoding human C-terminal peptides containing known or predicted Ser/Thr phosphosites and phosphomimetic variants thereof. We incorporated these oligonucleotides into a phage library and screened the PDZ (PSD-95/Dlg/ZO-1) domains of Scribble and DLG1 for interactions potentially enabled or disabled by ligand phosphorylation. We identified known and novel binders and characterized selected interactions through microscale thermophoresis, isothermal titration calorimetry, and NMR We uncover site-specific phospho-regulation of PDZ domain interactions, provide a structural framework for how PDZ domains accomplish phosphopeptide binding, and discuss ligand phosphorylation as a switching mechanism of PDZ domain interactions. The approach is readily scalable and can be used to explore the potential phospho-regulation of motif-based interactions on a large scale.
Keywords: PDZ domain; Scribble; phage display; phosphorylation; protein–protein interaction.
© 2018 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
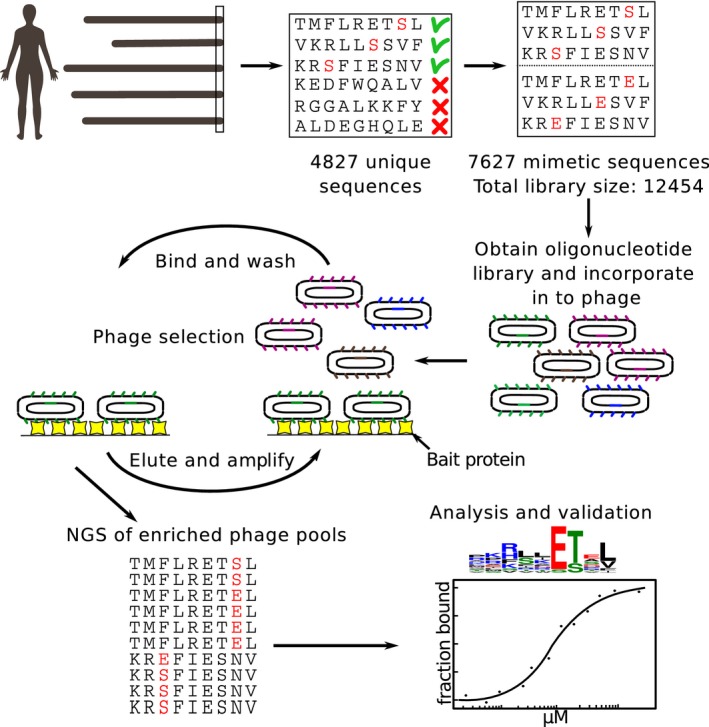
- A–D
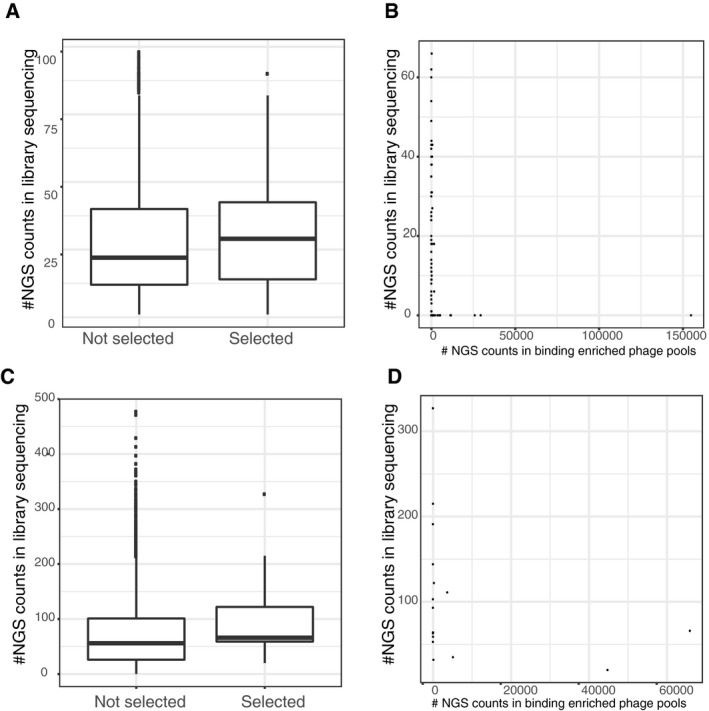
Upper panels (A and B): the phosphomimetic ProP‐PD experiment, lower panels (C and D): pre‐phosphorylation of the wild‐type library. Boxplots showing increased probability of a peptide being selected if being well represented in the initial library (y‐axis: read count in the respective initial library sequencing, peptides with no read support not used). Scatter plots show the read‐count after selection in the phage pools (x‐axis) against the read‐counts in the respective library sequencing (y‐axis).
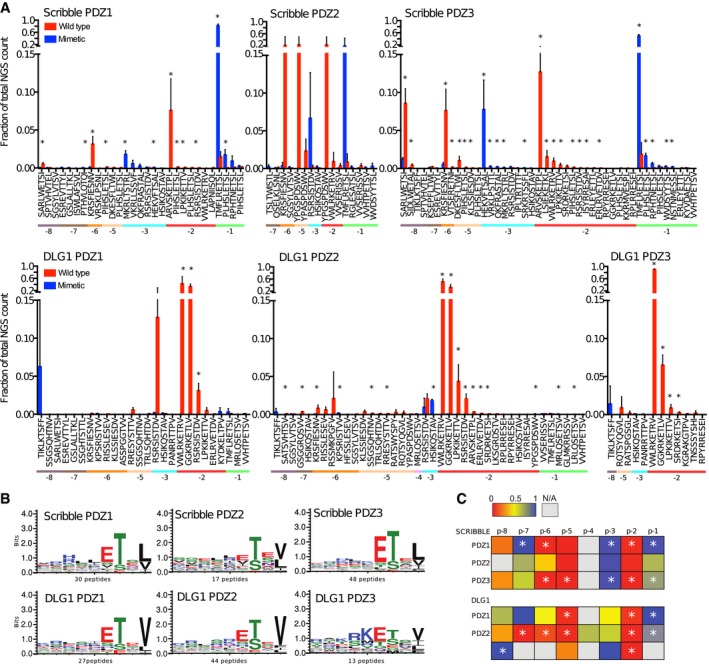
Fractions of the sequencing counts for individual peptides (wild‐type peptides indicated by red bars and phosphomimetic variants by blue bars) from NGS analysis of binding‐enriched phage pools from selections against the PDZ domains of Scribble and DLG1. Peptides are sorted based on the site of the phosphomimetic mutations (corresponding to known or putative phosphosites). Standard deviations are indicated (n = 3 for all cases except for Scribble PDZ2 where n = 2). Peptide pairs for which there are significant differences between the fractions of NGS counts for wild‐type and phosphomimetic peptides are indicated with * (multiple t‐tests, significance level 0.05). Peptides with multiple phosphorylation sites have been omitted from this analysis for clarity. For further details, see Table EV3.
Position weight matrices (PWMs, WebLogo3) representing the phosphomimetic ProP‐PD selections data for the PDZ domains of Scribble and DLG1. The numbers of peptides used for the analysis are indicated.
Scoring matrices of the effects of phosphomimetic mutations on the phosphomimetic ProP‐PD results of the PDZ domains of Scribble and DLG1. Scores are calculated from the ratios between NGS counts of the phosphomimetic peptides and the sum of the NGS counts of the wild‐type and phosphomimetic peptides. A score of 0 (red) indicates that the selection was dominated by wild‐type peptides, and a score of 1 (blue) indicates that the selection was dominated by peptides with phosphomimetic mutations at the given position (n = 3 for all cases except for Scribble PDZ2 where n = 2). Positions marked with * have a score that is significantly different from the score 0.5 of a neutral effect as determined using multiple t‐tests and correcting for multiple comparisons using a false discovery rate of 2.5%. The position for which only wild‐type or phosphomimetic peptides are represented in the data set could not be subjected to this test.
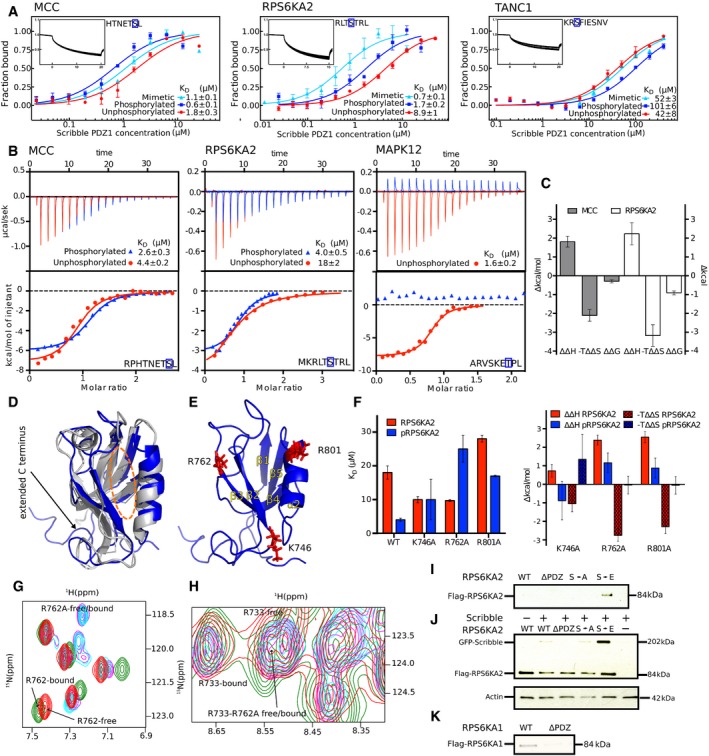
MST affinity measurements using FITC‐labeled peptides (unphosphorylated, phosphorylated, and phosphomimetic variants) of MCC (p‐1), RPS6KA2 (p‐3), and TANC1 (p‐6). A fixed peptide concentration (25–50 nM) was titrated with varying concentrations of Scribble PDZ1. K D values were determined using thermophoresis and T‐Jump signal for data analysis (n = 3; error bars represent SD). Statistical assessment using ordinary one‐way ANOVA for multiple comparisons confirmed significant differences (P ≤ 0.001) between the affinities for the phosphorylated peptides compared to the unphosphorylated peptides of MCC, RPS6KA2, and TANC1. For the comparison between the wild‐type and phosphomimetic peptides, there were significant differences for the peptides of MCC (P ≤ 0.01) and RPS6KA2 (P ≤ 0.001), but not for TANC1. Insets show representative titrations
Representative ITC titrations of Scribble PDZ1 with unphosphorylated and phosphorylated peptides of MCC, RPS6KA2, and MAPK12 (n = 3). Statistical assessment using multiple t‐tests corrected using the Holm–Sidak method for multiple comparisons show a significant difference between the phosphorylated and unphosphorylated MCC (P = 0.00098) and RPS6KA2 (P = 0.0003) peptides.
Differences between the thermodynamic parameters of Scribble PDZ1 when binding unphosphorylated or phosphorylated ligands (MCC and RPS6KA2) as determined in (B) (error bars represent SD, n = 3).
Overlay of the previously published unliganded structure of Scribble PDZ1 (gray; PDB code 1X5Q) and the here determined NMR structure (blue; PDB code 6ESP) of the protein bound to MKRLTpSTRL‐coo‐ (peptide not shown). The canonical PDZ binding grove is indicated by a dotted orange line.
Structure of the phosphopeptide‐bound Scribble PDZ1 showing three positively charged residues (K746, R762, and R801) surrounding the peptide binding pocket.
Left panel: Equilibrium binding constants for the binding between Scribble PDZ1 wild type and mutants (K746A, R762A, and R801A) and RPS6KA2 (unphosphorylated and phosphorylated) as determined through ITC titrations (n = 3). Right panel: Changes in the thermodynamic parameters upon mutation (ΔΔH mutation and −TΔΔS mutation) as determined through ITC (error bars represent SD, n = 3).
Section of the 15N‐1H HSQC spectra of ligand‐free wild‐type Scribble PDZ1 (red) and the protein bound to the phospho‐RPS6KA2 peptide (green), together with the spectra of the ligand‐free R762A mutant (magenta) and the mutant bound to phospho‐RPS6KA2 (cyan). Note that residue 762 experiences chemical shift perturbation only in the wild‐type Scribble PDZ1.
Slice of the 15N‐1H HSQC spectra showing the R733 or R762A residue of the wild‐type or mutant proteins. R733 is critical for mediating the carboxylate at the C‐terminus, and its position does not change in the wild‐type‐bound or the R762A‐bound form, showing that the carboxylate is still making the important interaction.
GST‐pulldown of full‐length Flag‐tagged RPS6KA2 wild type and mutants (S730A and S730E) and a truncated version (S730Δ) over‐expressed in HEK293T cells with GST‐tagged Scribble PDZ1. Detection was performed using an anti‐Flag antibody.
Co‐IPs of GFP‐tagged full‐length Scribble with Flag‐tagged full‐length RPS6KA2 constructs as indicated. Detection was performed using an anti‐Flag antibody and an anti‐GFP antibody.
GST‐pulldown of full‐length Flag‐tagged RPS6KA1 wild type and a truncated version of the protein (S732Δ). Detection was performed using an anti‐Flag antibody.
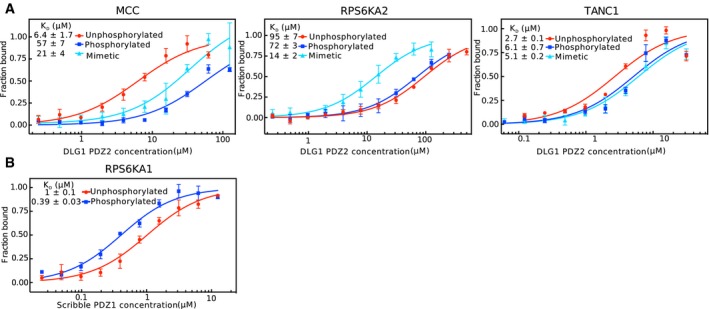
Titration of DLG1 PDZ2 to unphosphorylated, phosphorylated, or phosphomimetic variants of MCC (p‐1), RPS6KA2 (p‐3), and TANC1(p‐6).
Titration of Scribble PDZ1 to the unphosphorylated or p‐3 phosphorylated peptide of RPS6KA1 (VRKLPSTTL‐coo‐).
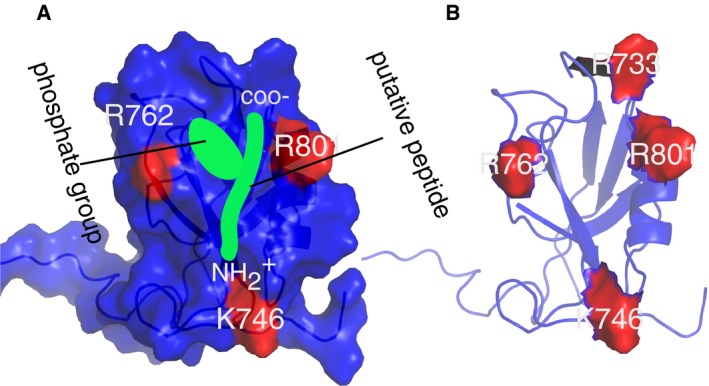
Surface representation of Scribble PDZ1 showing charge residues in the vicinity of the putative peptide (green). The carboxylate, amino terminal as well as the phosphate group of the peptide are indicated. Notice the cavity created by the phosphate‐group and the C‐terminus carboxylate.
Scribble PDZ1 showing all the positively charged residues surrounding the peptide binding site.
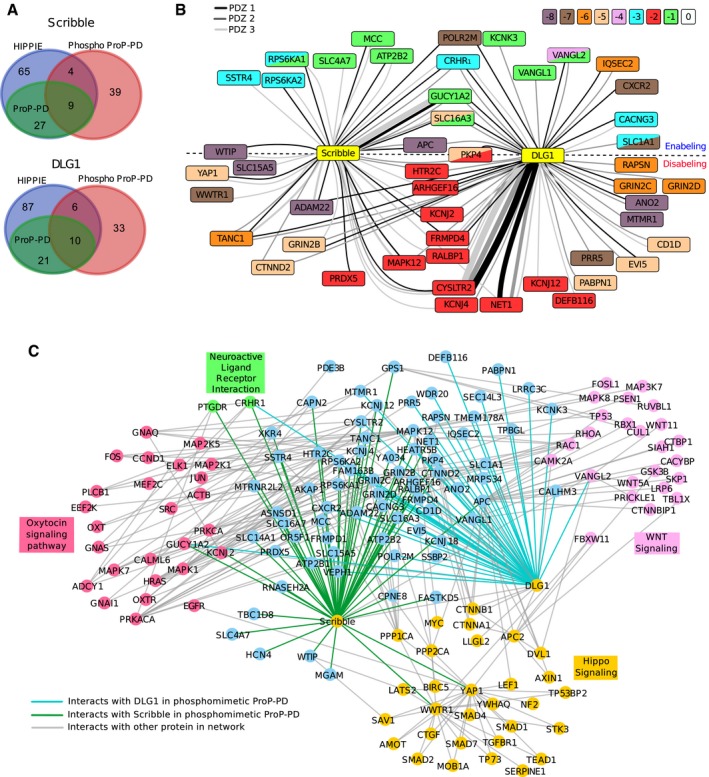
Overlap between ligands identified through phosphomimetic ProP‐PD (red) and interactions reported in the HIPPIE database (blue). The HIPPIE database already contains ligands previously reported through ProP‐PD (green; Ivarsson et al, 2014) for Scribble and DLG1.
Network representation of high‐confidence Scribble and DLG1 ligands. The node color indicates the position of the known or predicted phosphorylation site. Double mutants are indicated by two colors within one node. The edge color indicates the domain (PDZ1, PDZ2, PDZ3) that was found to interact with the target. The edge thickness reflects the total count of the peptide (WT and phosphomimetic) in the NGS analysis. For targets above the dotted line, the phosphomimetic mutations enable interactions. For targets below the dotted line, interactions are disabled by mutations. Interactions with targets on the dotted line are enabled or disabled depending on which PDZ domain they interact with.
Extended network containing interactions with STRING score over 0.8 to any target protein that are present in KEGG pathways enriched in the STRING‐based enrichment analysis.
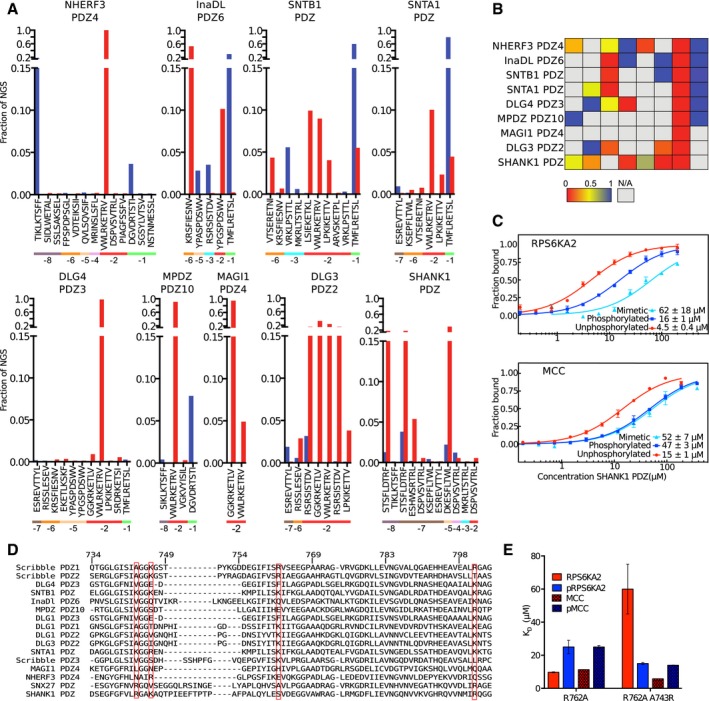
Phosphomimetic ProP‐PD selection results from selections against nine additional class I binding PDZ domains. The fractions of the sequencing counts from NGS analysis of the binding‐enriched phage pools for each peptide wild‐type and phosphomimetic peptide pair (wild type in red bars and phosphomimetic peptide in blue). The site of the phosphomimetic mutations is indicated below the peptide sequences. Peptides with multiple phosphorylation sites have been removed for clarity.
Scoring matrices of the effects of phosphomimetic mutations on the phosphomimetic ProP‐PD results of the PDZ domain scores are calculated from the ratios of NGS counts of the phosphomimetic peptides and the sum of the NGS counts of the wild‐type and phosphomimetic peptides. A score of 0 (red) indicates that the selection was dominated by wild‐type peptides, and a score of 1 (blue) indicates that the selection was dominated by peptides with phosphomimetic mutations at the given position.
MST affinity measurements using FITC‐labeled peptides (unphosphorylated, phosphorylated, and phosphomimetic) of MCC (p‐1) and RPS6KA2 (p‐3) and SHANK1 PDZ. A fixed peptide concentration (25–50 nM) was titrated with varying concentrations of the protein. K D values were determined using thermophoresis and T‐Jump signal for data analysis (n = 3; error bars represent SD). Statistical assessment using ordinary one‐way ANOVA for multiple comparisons confirmed significant affinity differences between wild‐type and phosphomimetic RPS6KA2 (P ≤ 0.01) and unphosphorylated and phosphomimetic or phosphorylated MCC (P ≤ 0.001).
Structure‐based sequence alignment of the PDZ domains explored in this study and SNX27 PDZ that was recently shown to bind phosphopeptides (Clairfeuille et al, 2016). The numbering is based on Scribble, and the sequences are ordered based on sequence similarities with Scribble PDZ1. The red boxes indicate the basic residues of Scribble PDZ1 mutated in Fig 3, and residue A743, which aligns with the R58 of the SNX27 PDZ domain that was recently described to be of importance for binding p‐3 and p‐5 phosphorylated ligands.
Histogram of equilibrium dissociation constants of Scribble PDZ1 R762A (mutational background) and the R762A/A743R mutant with unphosphorylated and phosphorylated MCC and RPS6KA2 peptides (error bars represent the SD, n = 3). Note that the A743R mutation restores the preference for the p‐3 phosphorylated RPS6KA2 peptide.
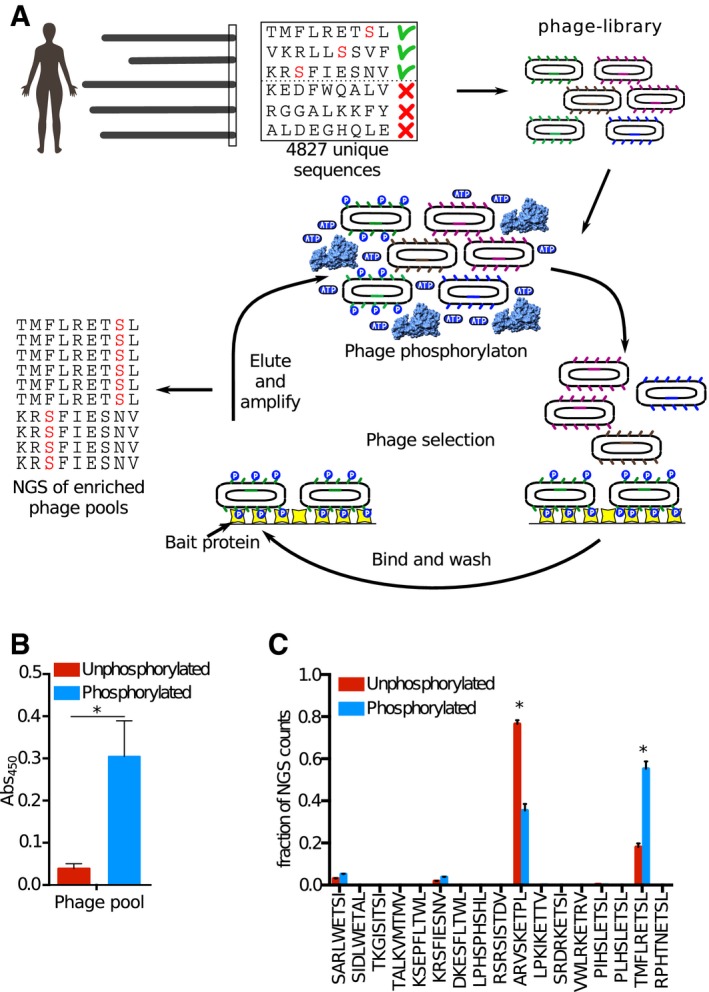
We constructed a ProP‐PD library that displays peptides that represent the C‐terminal peptides of the human proteome with known or putative Ser/Thr phosphorylation sites (i.e., the wild‐type part of the phosphomimetic ProP‐PD library design). The library was subjected to phosphorylation by RPS6KA1 prior to each round of selection against the bait Scribble PDZ1. The binding‐enriched phage pools were analyzed by NGS, and the results were compared to the results of a selection against the same library but without phosphorylation.
Phosphorylation of the ProP‐PD library was confirmed through sandwich ELISA. An anti‐phospho‐Ser/Thr/Tyr antibody was immobilized and challenged with the phosphorylated or unphosphorylated ProP‐PD library, and the binding was detected using an HRP‐conjugated anti‐M13 antibody (error bars represent SD, n = 2). A significant difference in the binding to the phospho‐antibody was observed for the phosphorylated ProP‐PD library as compared to unphosphorylated library (*P ≤ 0.05, unpaired t‐test).
Bar graphs representing the fraction of NGS counts for distinct peptides from the selections against the pre‐phosphorylated and unphosphorylated libraries (error bars represent SD, n = 2). For peptides indicated with *, there are significant differences between the fractions of NGS counts from the distinct selections, using multiple t‐tests.
References
-
- Adey NB, Huang L, Ormonde PA, Baumgard ML, Pero R, Byreddy DV, Tavtigian SV, Bartel PL (2000) Threonine phosphorylation of the MMAC1/PTEN PDZ binding domain both inhibits and stimulates PDZ binding. Can Res 60: 35–37 - PubMed
-
- Blom N, Sicheritz‐Ponten T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post‐translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4: 1633–1649 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
