

Jujuboside A promotes Aβ clearance and ameliorates cognitive deficiency in Alzheimer's disease through activating Axl/HSP90/PPARγ pathway
- PMID: 30128052
- PMCID: PMC6096387
- DOI: 10.7150/thno.26164
Jujuboside A promotes Aβ clearance and ameliorates cognitive deficiency in Alzheimer's disease through activating Axl/HSP90/PPARγ pathway
Abstract
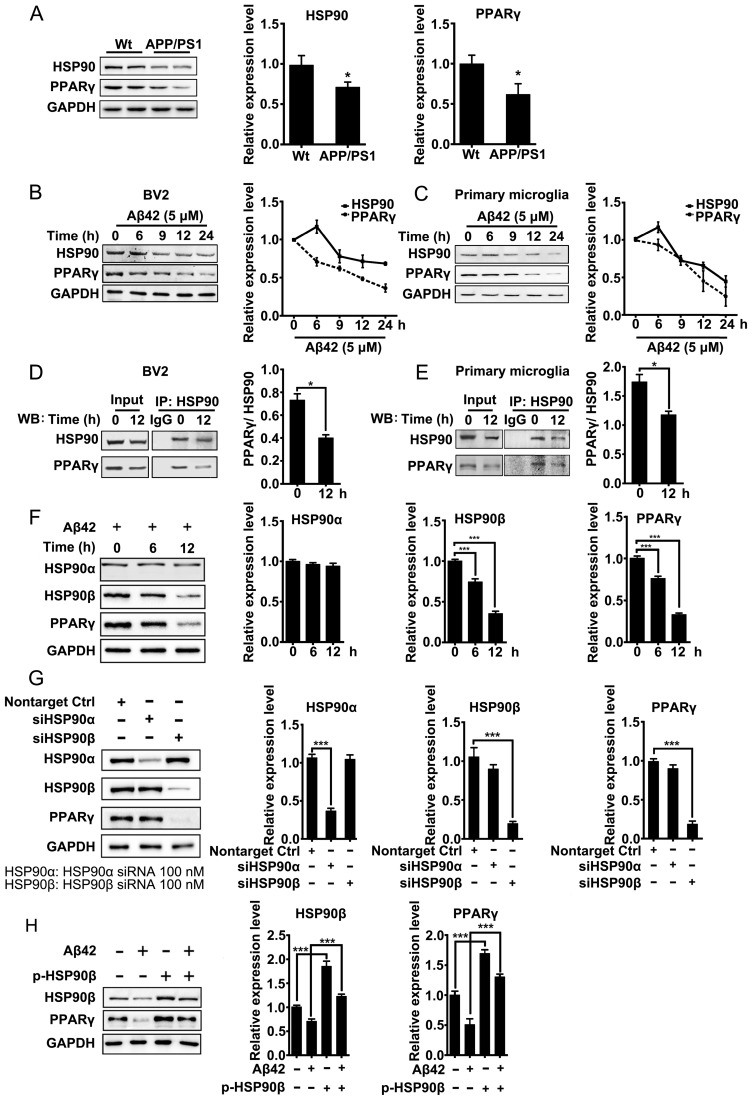
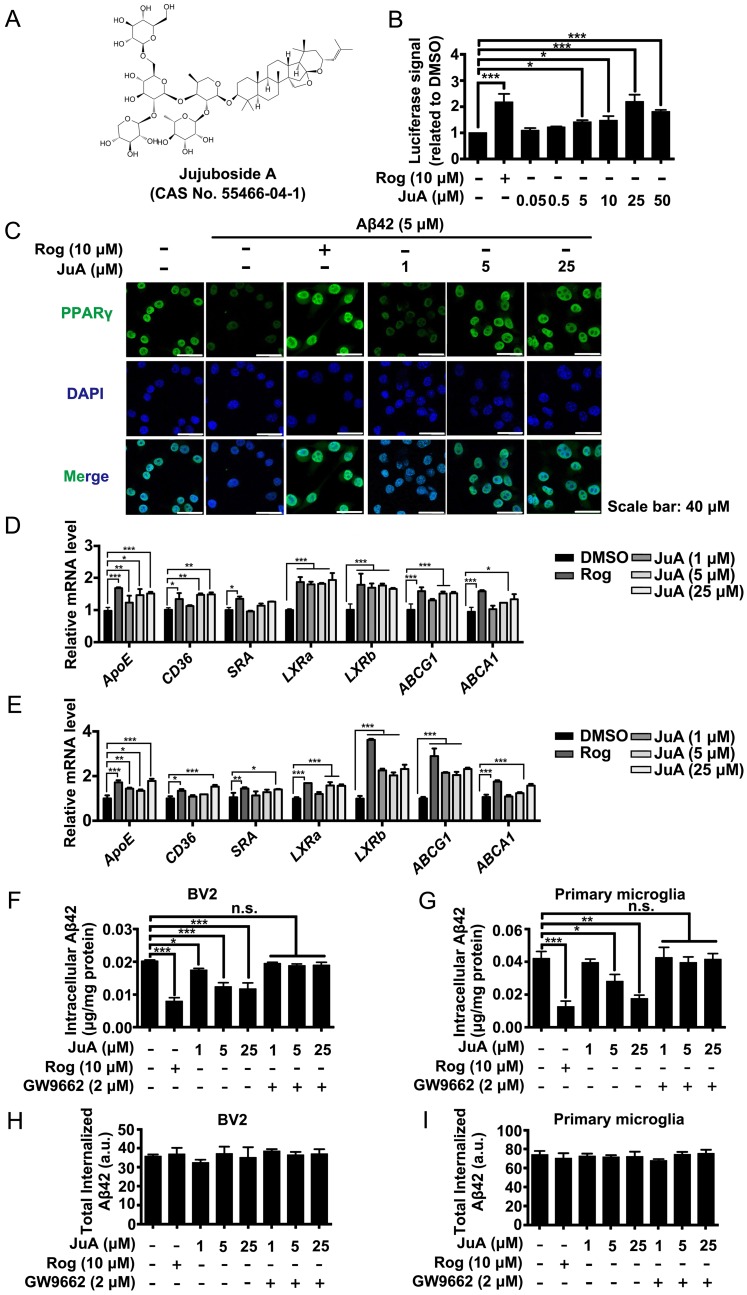
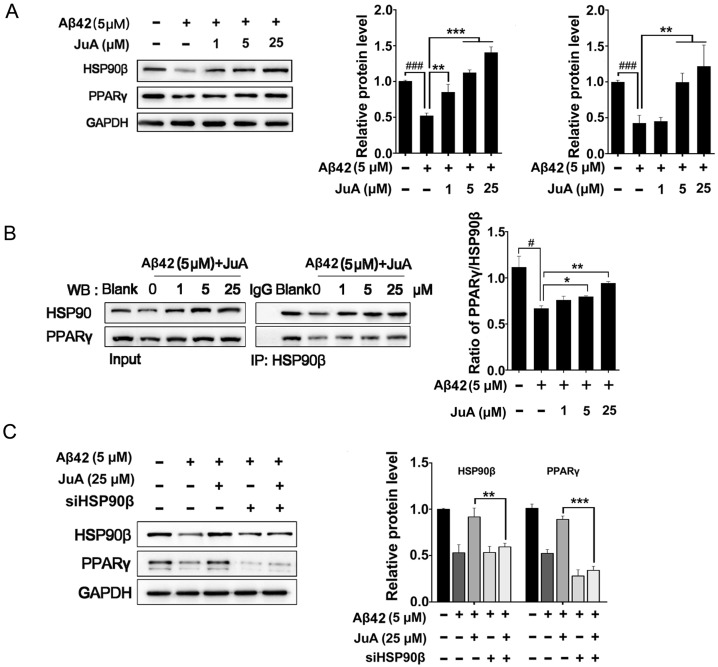
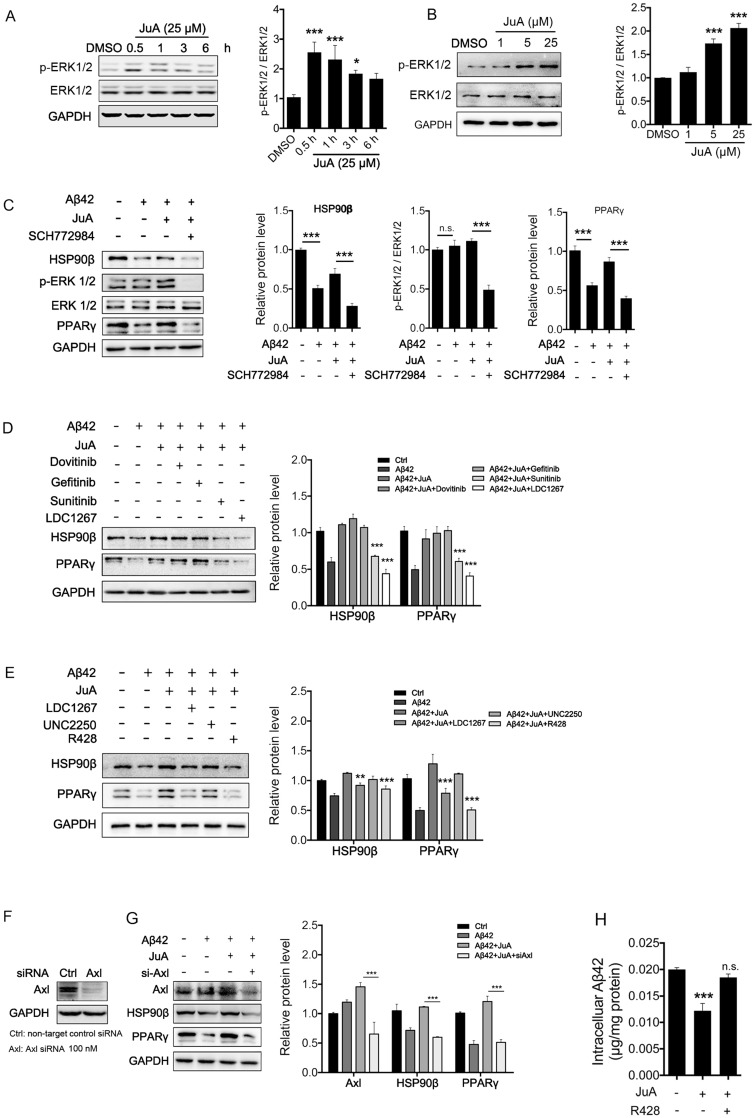
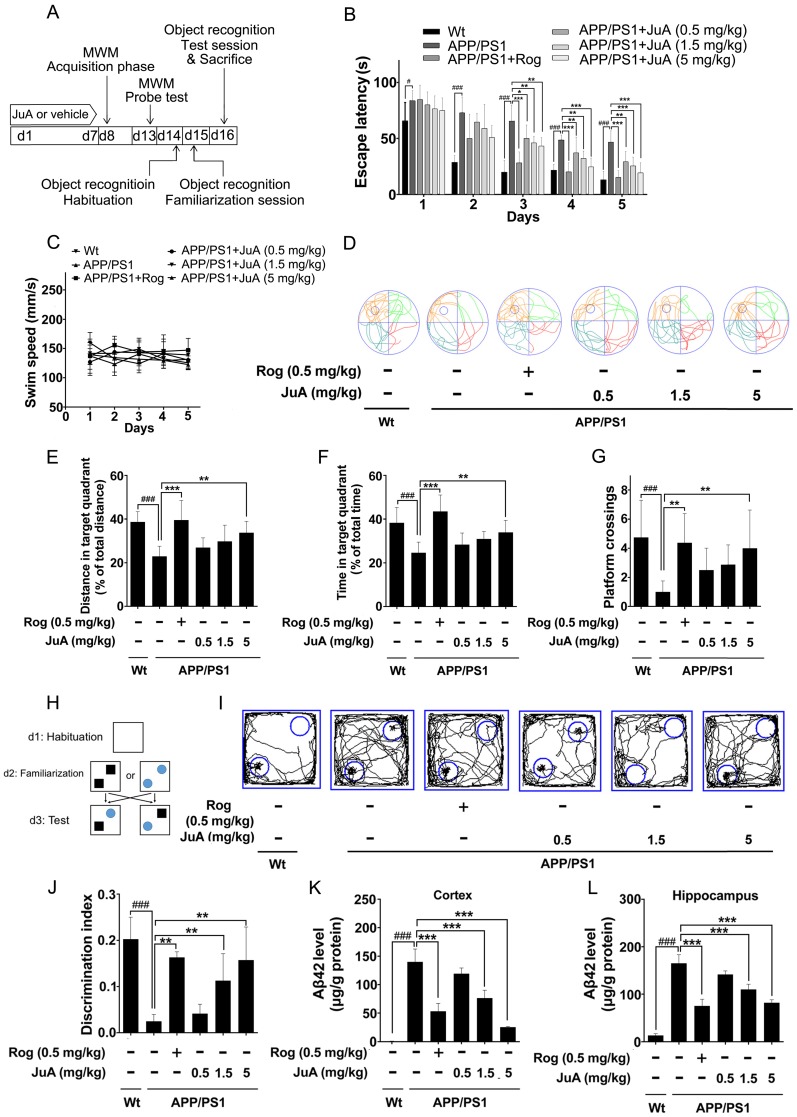
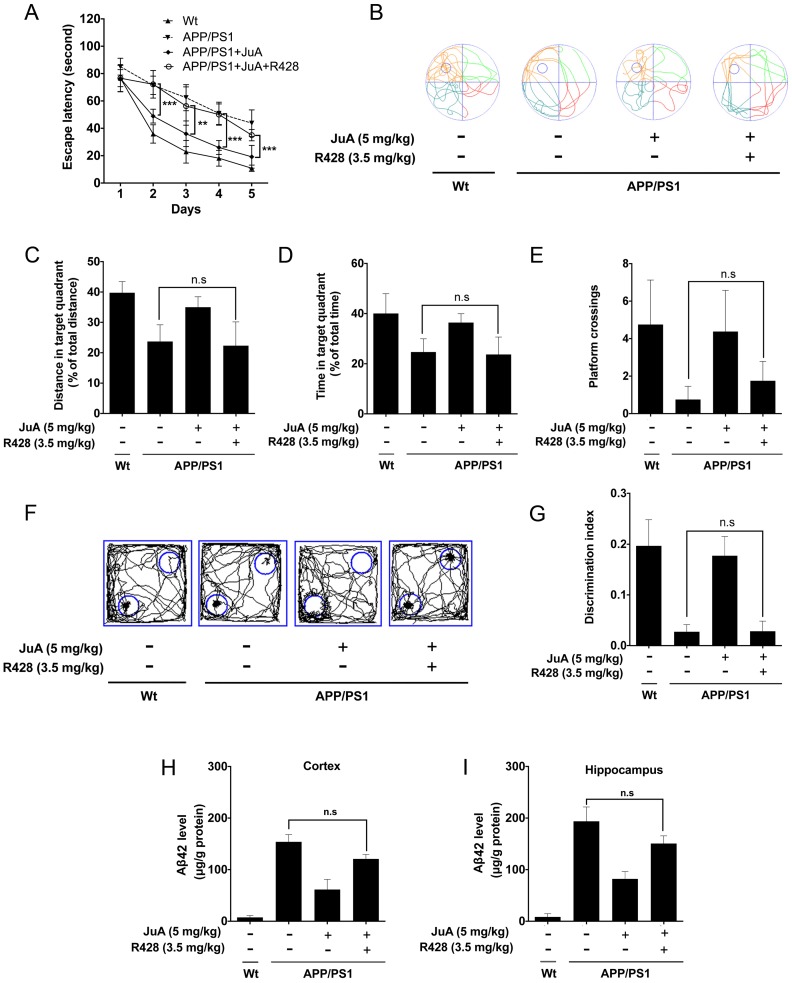
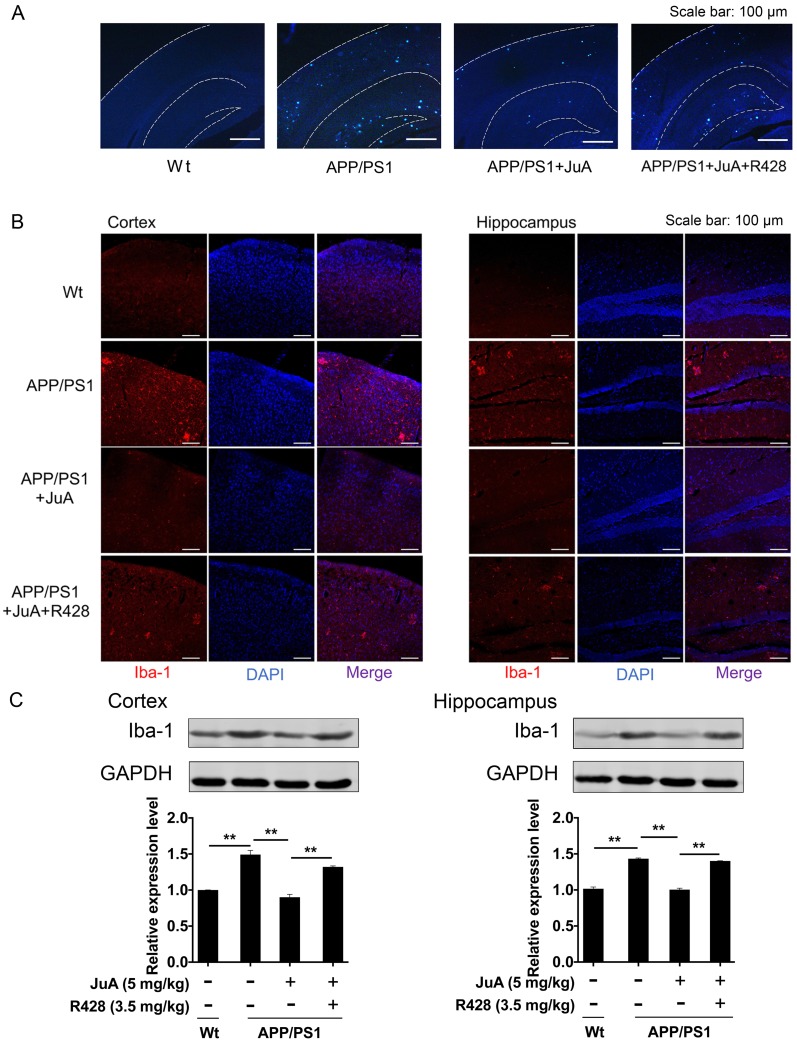
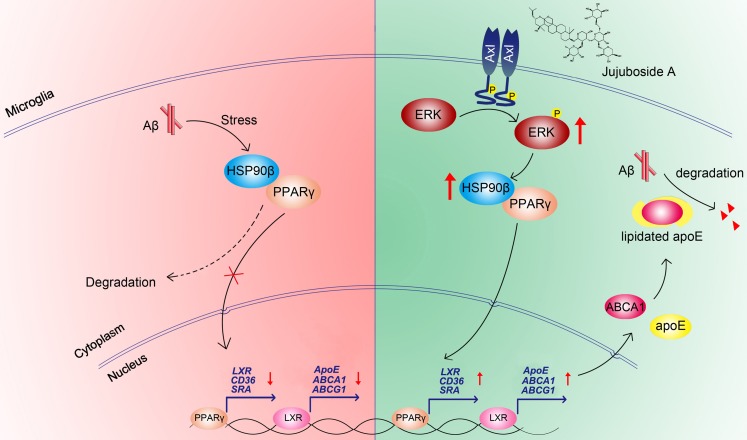
Rationale: It has been reported that peroxisome proliferator activated receptor γ (PPARγ) level decreases significantly in the brains of Alzheimer's disease (AD) patients and mice models, while the mechanism is unclear. This study aims to unravel the mechanism that amyloid β (Aβ) decreases PPARγ and attempted to discover lead compound that preserves PPARγ. Methods: In APP/PS1 transgenic mice and Aβ treated microglia, the interaction between HSP90 and PPARγ were analyzed by western blot. Using a PPRE (PPARγ responsive element) containing reporter cell line, compounds that activate PPARγ activity were identified. After genetic ablation or pharmacological inhibition of potential target pathways, the target of jujuboside A (JuA) was discovered through Axl/HSP90β. After oral administration or intrathecal injection, the anti-AD activity of JuA was evaluated by Morris water maze (MWM) test and object recognition test. Soluble Aβ42 levels and plaque numbers after JuA treatment were detected by thioflavin S staining, and the activation of microglia was assayed by immunofluorescence staining against Iba-1. Results: We found that Aβ stress decreased heat shock protein 90 β (HSP90β), subsequently reduced the abundance of PPARγ, and down-regulated Aβ clearance-related genes in BV2 cells and primary microglia. We identified that JuA stimulated the expression of HSP90β, strengthened the interaction between HSP90β and PPARγ, preserved PPARγ levels, and thus effectively promoted the clearance of Aβ42. We demonstrated that JuA increased HSP90β expression through Axl/ERK pathway. JuA significantly ameliorated cognitive deficiency in APP/PS1 transgenic mice, meanwhile, JuA significantly reduced the soluble Aβ42 levels and plaque numbers in the brain. Notably, the therapeutic effects of JuA were dampened by R428, an Axl inhibitor. Conclusions: This study suggests that the up-regulation of HSP90β by JuA through Axl is a potential therapeutic strategy to facilitate Aβ42 clearance and ameliorate cognitive deficiency in AD.
Keywords: Alzheimer's disease; Axl; HSP90β; Jujuboside A; PPARγ; amyloid β.
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. - PubMed
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. - PubMed
-
- Polanco JC, Li C, Bodea LG, Martinez-Marmol R, Meunier FA, Gotz J. Amyloid-beta and tau complexity - towards improved biomarkers and targeted therapies. Nat Rev Neurol. 2018;14:22–39. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
