

Simulations of lipid bilayers using the CHARMM36 force field with the TIP3P-FB and TIP4P-FB water models
- PMID: 30128211
- PMCID: PMC6097494
- DOI: 10.7717/peerj.5472
Simulations of lipid bilayers using the CHARMM36 force field with the TIP3P-FB and TIP4P-FB water models
Abstract
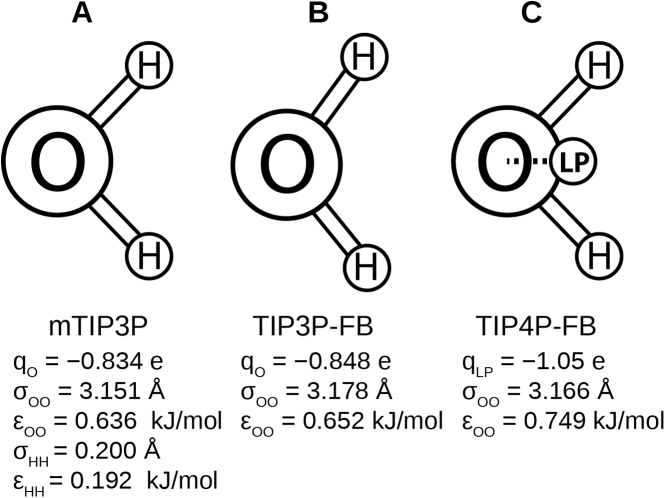
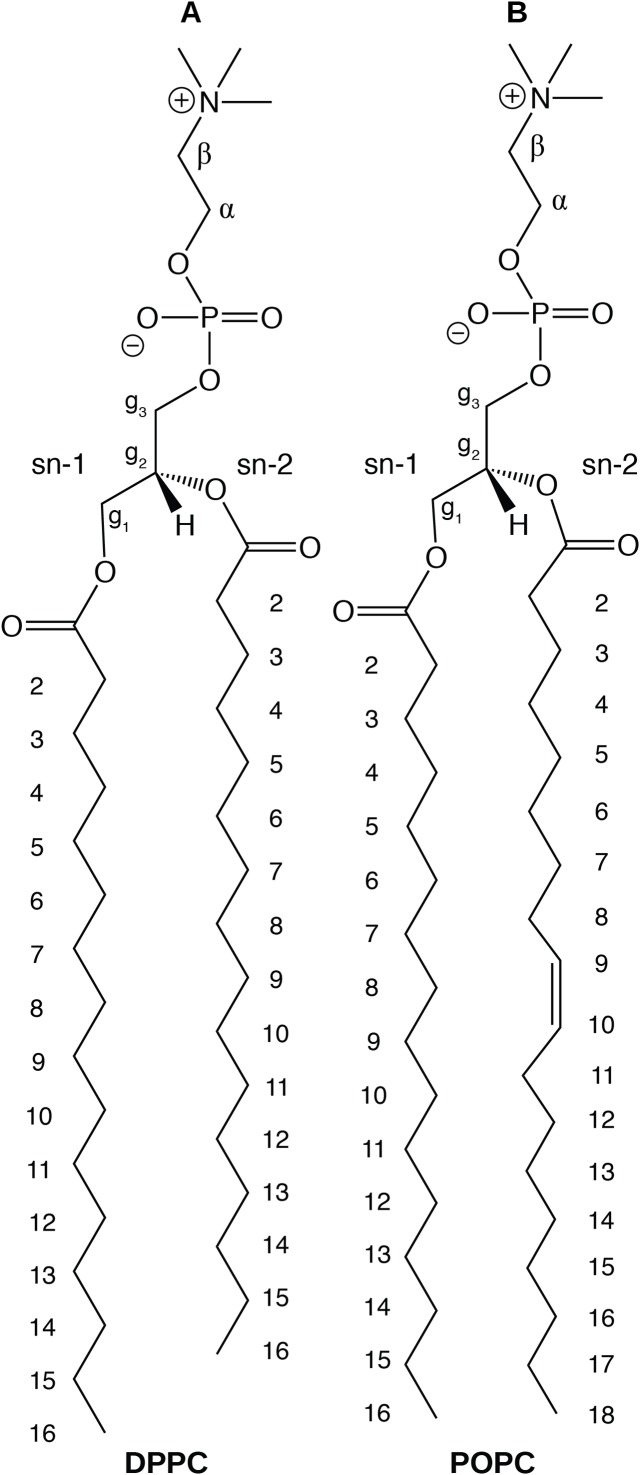
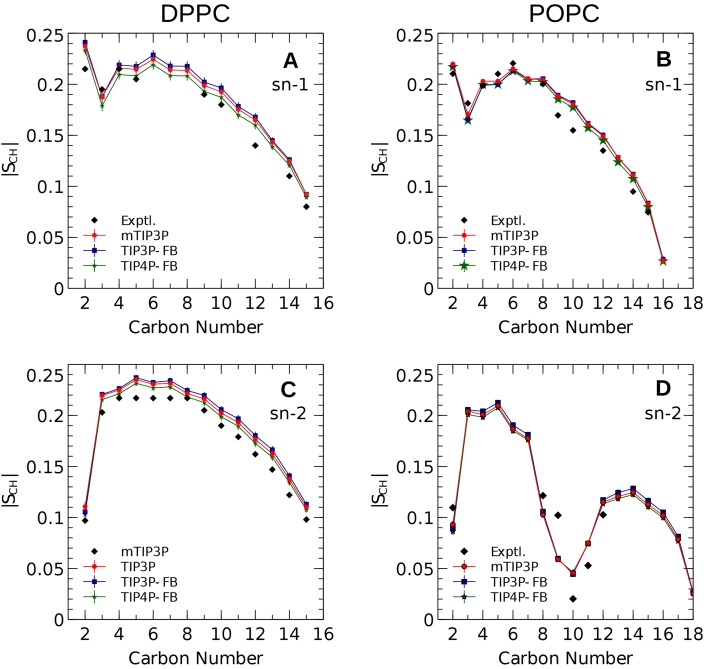
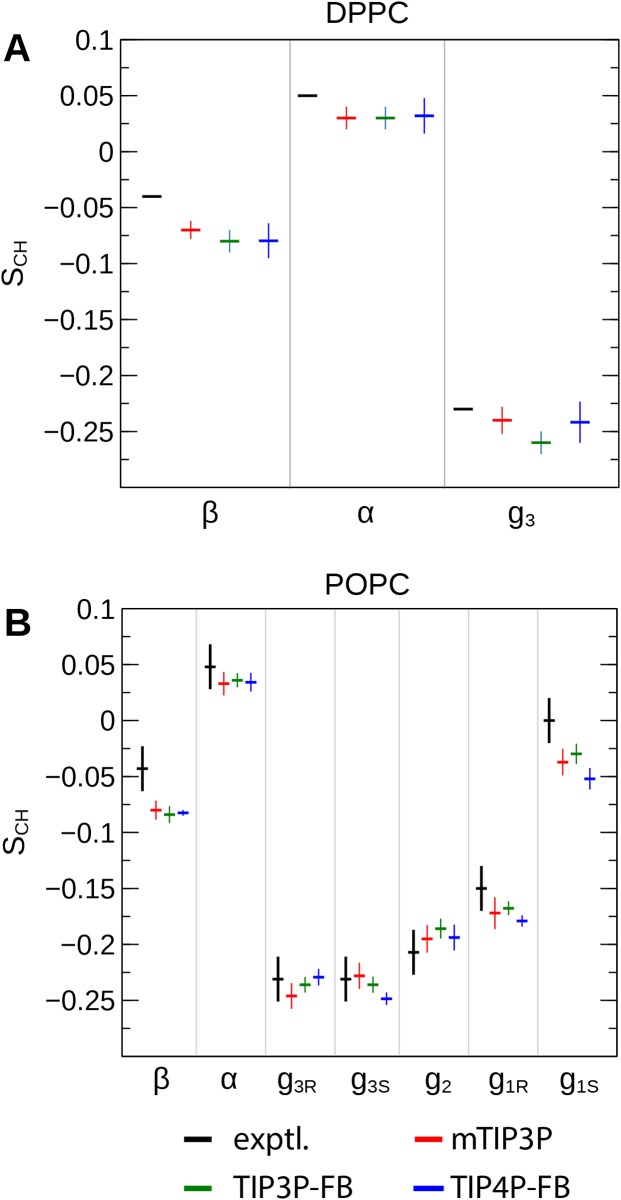
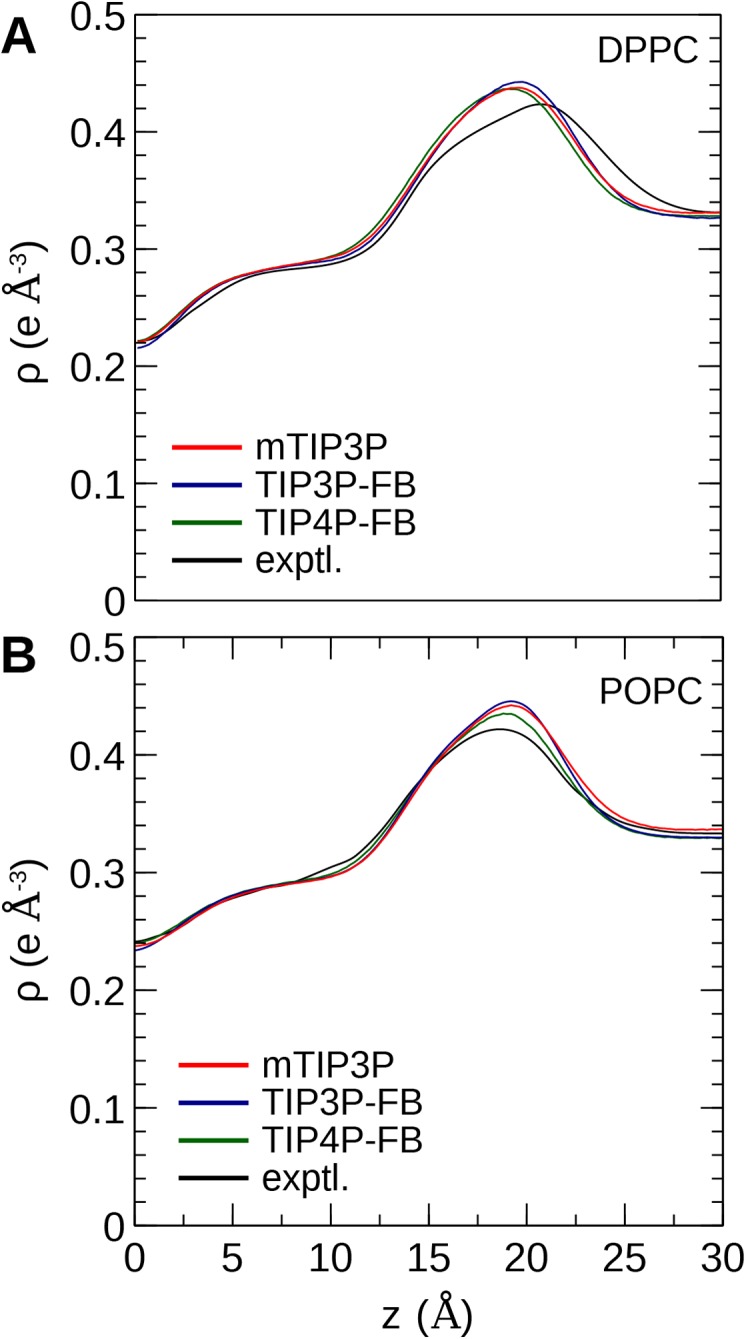
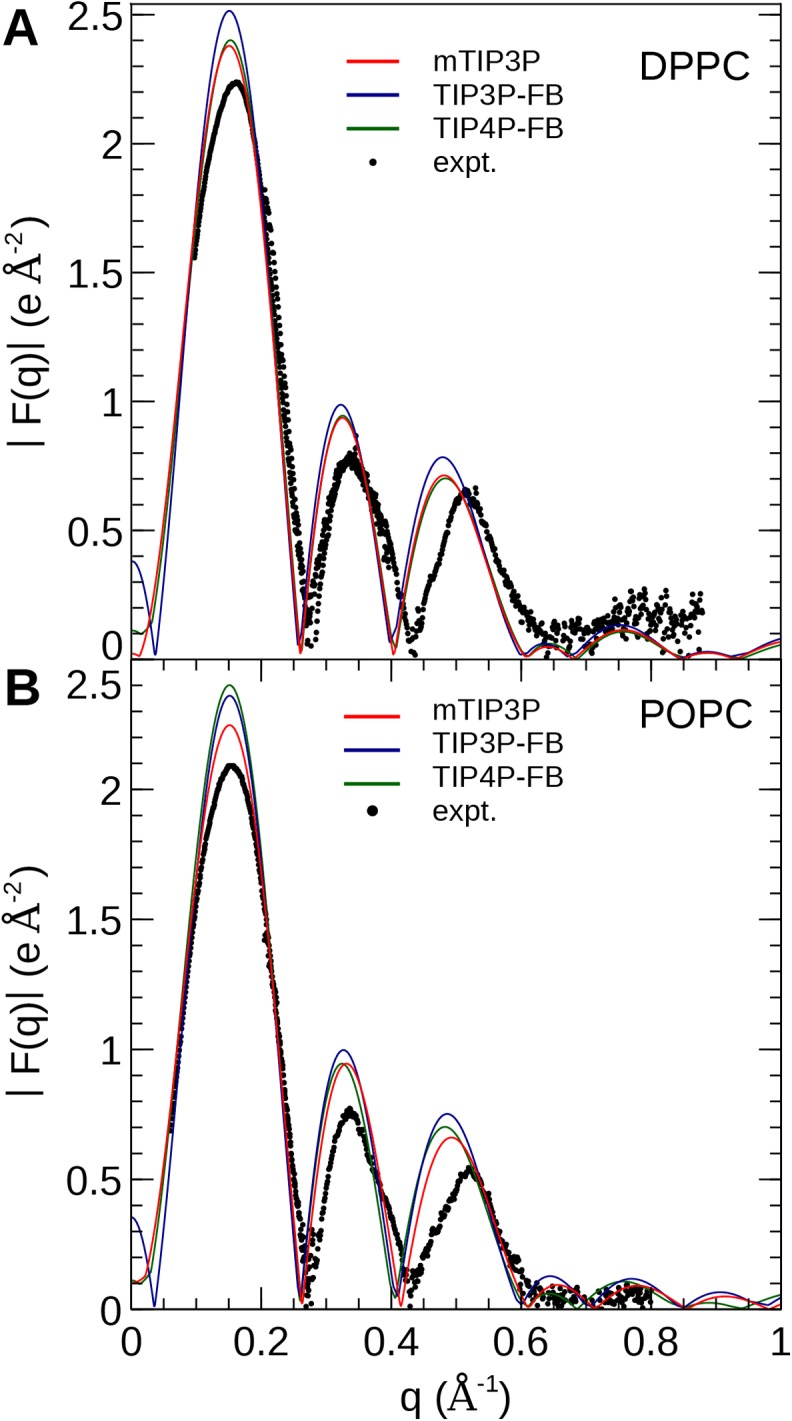
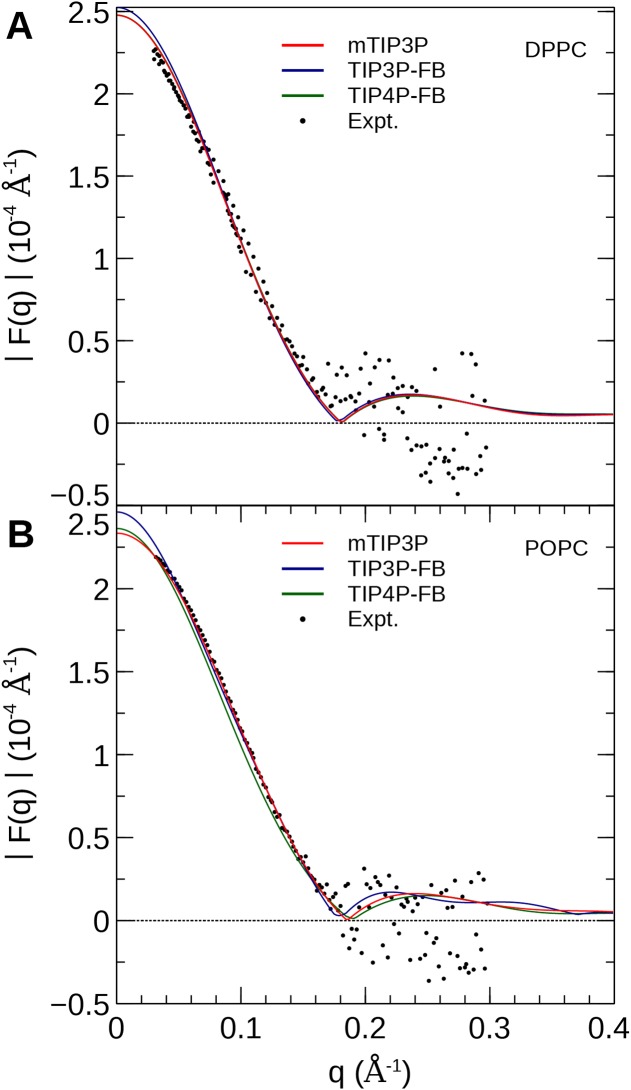
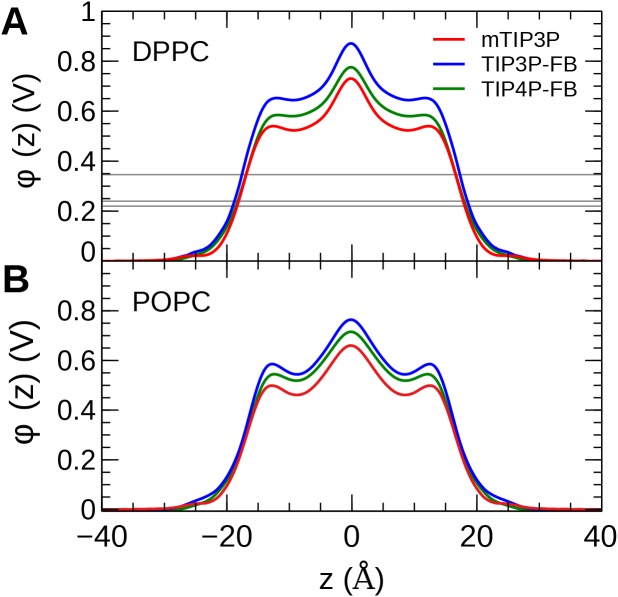
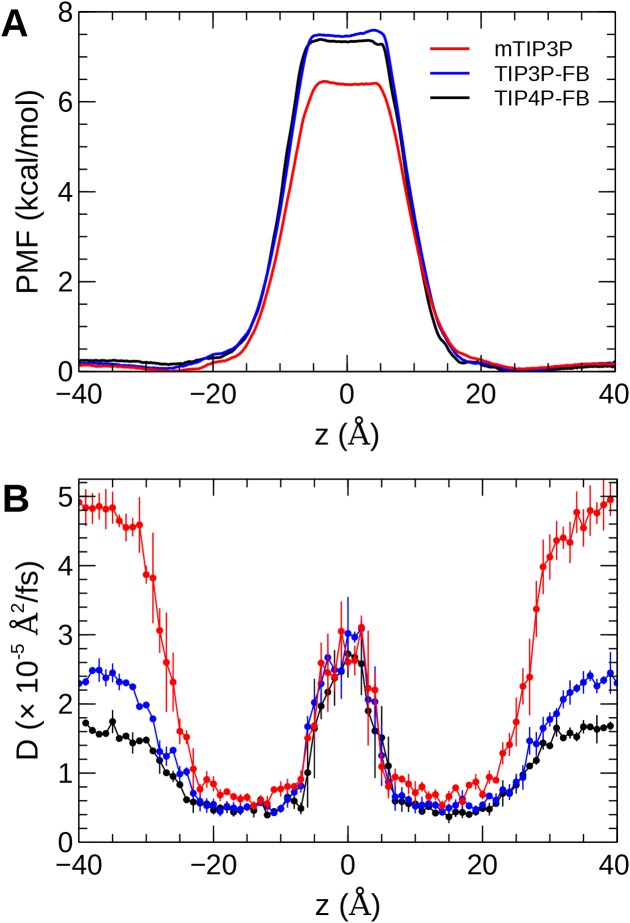
The CHARMM36 force field for lipids is widely used in simulations of lipid bilayers. The CHARMM family of force fields were developed for use with the mTIP3P water model. This water model has an anomalously high dielectric constant and low viscosity, which limits its accuracy in the calculation of quantities like permeability coefficients. The TIP3P-FB and TIP4P-FB water models are more accurate in terms of the dielectric constant and transport properties, which could allow more accurate simulations of systems containing water and lipids. To test whether the CHARMM36 lipid force field is compatible with the TIP3P-FB and TIP4P-FB water models, we have performed simulations of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayers. The calculated headgroup area, compressibility, order parameters, and X-ray form factors are in good agreement with the experimental values, indicating that these improved water models can be used with the CHARMM36 lipid force field without modification when calculating membrane physical properties. The water permeability predicted by these models is significantly different; the mTIP3P-model diffusion in solution and at the lipid-water interface is anomalously fast due to the spuriously low viscosity of mTIP3P-model water, but the potential of mean force of permeation is higher for the TIP3P-FB and TIP4P-FB models due to their high excess chemical potentials. As a result, the rates of water permeation calculated the FB water models are slower than the experimental value by a factor of 15-17, while simulations with the mTIP3P model only underestimate the water permeability by a factor of 3.
Keywords: Force field; Head group area; Lipid; Membrane permeability; Molecular dynamics; Molecular simulation; Scattering; Water model.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
CHARMM TIP3P Water Model Suppresses Peptide Folding by Solvating the Unfolded State.J Phys Chem B. 2016 Apr 21;120(15):3692-8. doi: 10.1021/acs.jpcb.6b01316. Epub 2016 Apr 12. J Phys Chem B. 2016. PMID: 27031562
-
Interactions of Water and Alkanes: Modifying Additive Force Fields to Account for Polarization Effects.J Chem Theory Comput. 2019 Jun 11;15(6):3854-3867. doi: 10.1021/acs.jctc.9b00016. Epub 2019 May 9. J Chem Theory Comput. 2019. PMID: 31002505 Free PMC article.
-
Force Field Benchmark of Amino Acids: I. Hydration and Diffusion in Different Water Models.J Chem Inf Model. 2018 May 29;58(5):1037-1052. doi: 10.1021/acs.jcim.8b00026. Epub 2018 Apr 18. J Chem Inf Model. 2018. PMID: 29648448
-
Polyphilic Interactions as Structural Driving Force Investigated by Molecular Dynamics Simulation (Project 7).Polymers (Basel). 2017 Sep 14;9(9):445. doi: 10.3390/polym9090445. Polymers (Basel). 2017. PMID: 30965747 Free PMC article. Review.
-
Shedding light on the structural properties of lipid bilayers using molecular dynamics simulation: a review study.RSC Adv. 2019 Feb 6;9(8):4644-4658. doi: 10.1039/c8ra08441f. eCollection 2019 Jan 30. RSC Adv. 2019. PMID: 35520151 Free PMC article. Review.
Cited by
-
Design, Screening, and Testing of Non-Rational Peptide Libraries with Antimicrobial Activity: In Silico and Experimental Approaches.Antibiotics (Basel). 2020 Nov 30;9(12):854. doi: 10.3390/antibiotics9120854. Antibiotics (Basel). 2020. PMID: 33265897 Free PMC article. Review.
-
Identification of a potent and selective LAPTc inhibitor by RapidFire-Mass Spectrometry, with antichagasic activity.PLoS Negl Trop Dis. 2024 Feb 15;18(2):e0011956. doi: 10.1371/journal.pntd.0011956. eCollection 2024 Feb. PLoS Negl Trop Dis. 2024. PMID: 38359089 Free PMC article.
-
Cryptotanshinone-Induced Permeabilization of Model Phospholipid Membranes: A Biophysical Study.Membranes (Basel). 2024 May 21;14(6):118. doi: 10.3390/membranes14060118. Membranes (Basel). 2024. PMID: 38921485 Free PMC article.
-
Structure-Based Virtual Screening Reveals Ibrutinib and Zanubrutinib as Potential Repurposed Drugs against COVID-19.Int J Mol Sci. 2021 Jun 30;22(13):7071. doi: 10.3390/ijms22137071. Int J Mol Sci. 2021. PMID: 34209188 Free PMC article.
-
Molecular Dynamics Simulations of Rhodamine B Zwitterion Diffusion in Polyelectrolyte Solutions.J Phys Chem B. 2022 Dec 8;126(48):10256-10272. doi: 10.1021/acs.jpcb.2c06281. Epub 2022 Nov 28. J Phys Chem B. 2022. PMID: 36440862 Free PMC article.
References
-
- Abraham MH, Whiting GS, Fuchs R, Chambers EJ. Thermodynamics of solute transfer from water to hexadecane. Journal of the Chemical Society, Perkin Transactions 2. 1990;2:291–300. doi: 10.1039/p29900000291. - DOI
-
- Allen M, Tildesley D. Computer Simulation of Liquids. Oxford: Oxford Science Publications, Clarendon Press; 1989.
-
- Ben-Naim A, Marcus Y. Solvation thermodynamics of nonionic solutes. Journal of Chemical Physics. 1984;81(4):2016–2027. doi: 10.1063/1.447824. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
