

A review of clinical characteristics and genetic backgrounds in Alport syndrome
- PMID: 30128941
- PMCID: PMC6510800
- DOI: 10.1007/s10157-018-1629-4
A review of clinical characteristics and genetic backgrounds in Alport syndrome
Abstract
Alport syndrome (AS) is a progressive hereditary renal disease that is characterized by sensorineural hearing loss and ocular abnormalities. It is divided into three modes of inheritance, namely, X-linked Alport syndrome (XLAS), autosomal recessive AS (ARAS), and autosomal dominant AS (ADAS). XLAS is caused by pathogenic variants in COL4A5, while ADAS and ARAS are caused by those in COL4A3/COL4A4. Diagnosis is conventionally made pathologically, but recent advances in comprehensive genetic analysis have enabled genetic testing to be performed for the diagnosis of AS as first-line diagnosis. Because of these advances, substantial information about the genetics of AS has been obtained and the genetic background of this disease has been revealed, including genotype-phenotype correlations and mechanisms of onset in some male XLAS cases that lead to milder phenotypes of late-onset end-stage renal disease (ESRD). There is currently no radical therapy for AS and treatment is only performed to delay progression to ESRD using nephron-protective drugs. Angiotensin-converting enzyme inhibitors can remarkably delay the development of ESRD. Recently, some new drugs for this disease have entered clinical trials or been developed in laboratories. In this article, we review the diagnostic strategy, genotype-phenotype correlation, mechanisms of onset of milder phenotypes, and treatment of AS, among others.
Keywords: ACE inhibitor; Bardoxolone; Genotype–phenotype correlation; Thin basement membrane.
Conflict of interest statement
Conflict of interest
Kazumoto Iijima has received grant support from Daiichi Sankyo Co. Ltd. He has also received consulting fees from Takeda Pharmaceutical Company and Kyowa Hakko Kirin Co. Ltd. Kandai Nozu has received lecture fees from Novartis Pharmaceuticals Corporation. Kazumoto Iijima and Kandai Nozu have filed a patent application on the development of antisense nucleotides for exon skipping therapy in Alport syndrome. The remaining authors have declared that no conflict of interest exists.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was not required, as this study did not involve human participants.
Figures


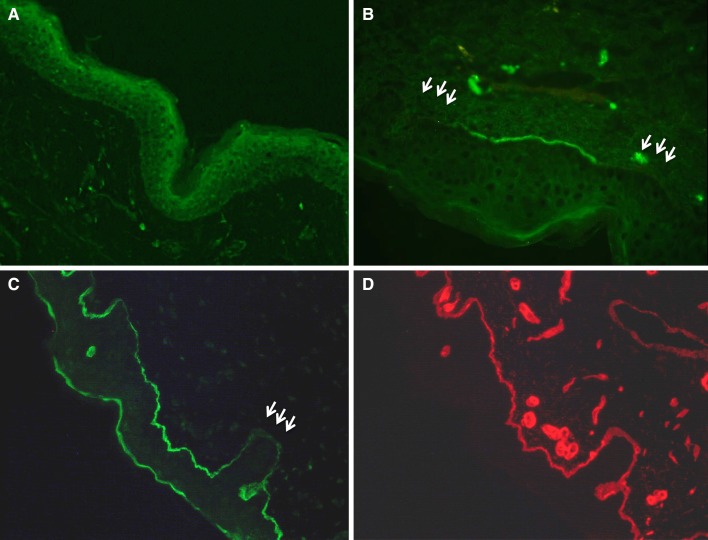

References
-
- Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine (Baltimore) 1999;78(5):338–360. - PubMed
-
- Nakanishi K, Yoshikawa N, Iijima K, Kitagawa K, Nakamura H, Ito H, et al. Immunohistochemical study of alpha 1–5 chains of type IV collagen in hereditary nephritis. Kidney Int. 1994;46(5):1413–1421. - PubMed
-
- Rheault MN, Kashtan CE. Inherited glomerular diseases. Ped Nephrol. 2016;7:191884.
-
- Mallett A, Tang W, Clayton PA, Stevenson S, McDonald SP, Hawley CM, et al. End-stage kidney disease due to Alport syndrome: outcomes in 296 consecutive Australia and New Zealand dialysis and transplant registry cases. Nephrol Dial Transplant. 2014;29(12):2277–2286. - PubMed
-
- Hattori M, Sako M, Kaneko T, Ashida A, Matsunaga A, Igarashi T, et al. End-stage renal disease in Japanese children: a nationwide survey during 2006–2011. Clin Exp Nephrol. 2015;19(5):933–938. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
