

Interferon at the cellular, individual, and population level in hepatitis C virus infection: Its role in the interferon-free treatment era
- PMID: 30129199
- PMCID: PMC6614028
- DOI: 10.1111/imr.12689
Interferon at the cellular, individual, and population level in hepatitis C virus infection: Its role in the interferon-free treatment era
Abstract
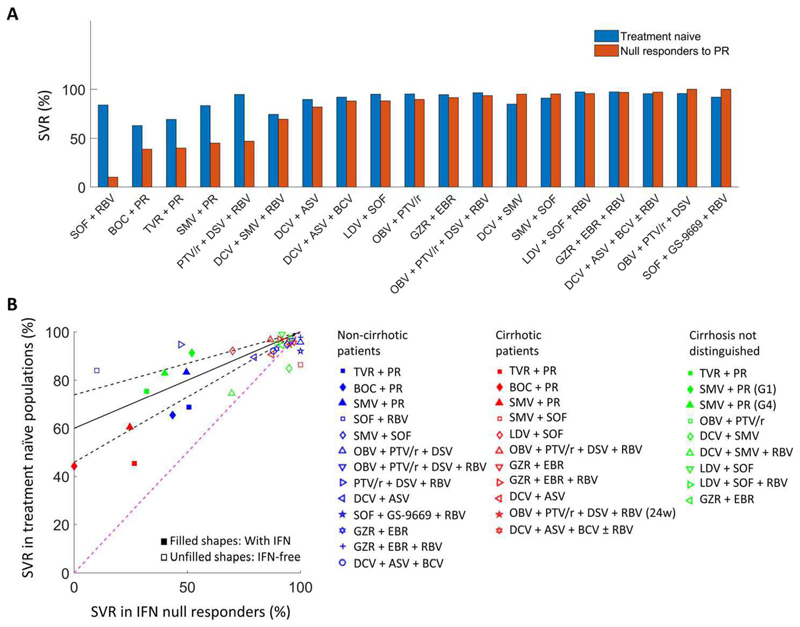
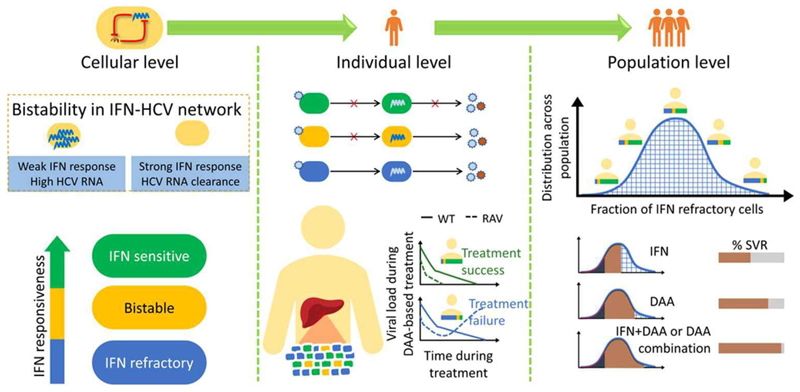
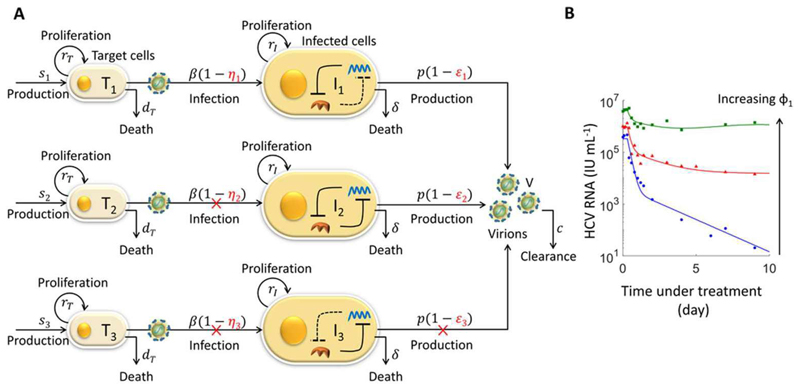
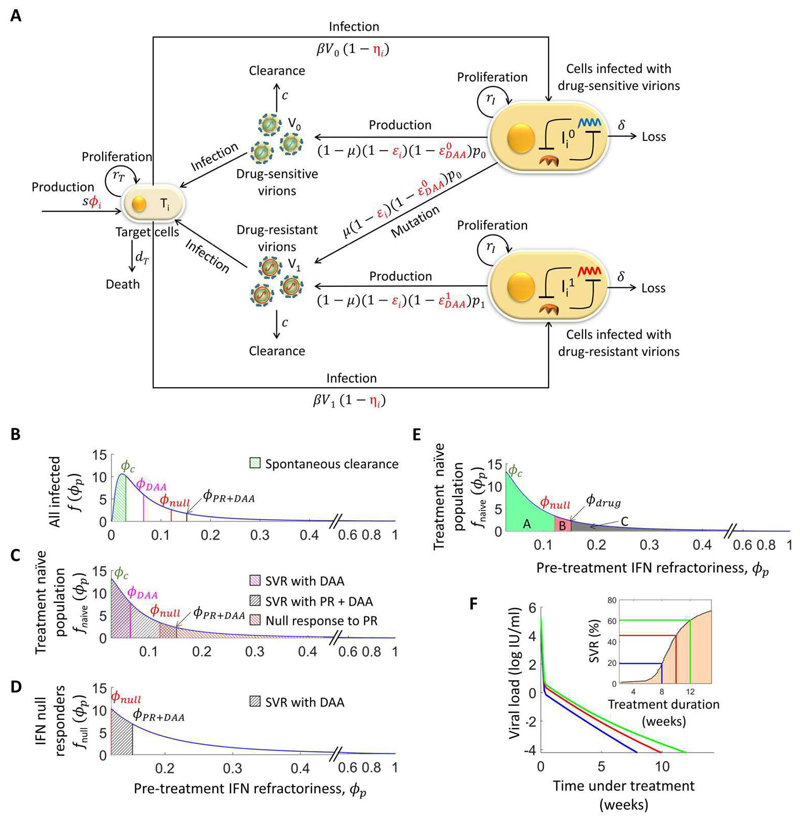
The advent of powerful direct-acting antiviral agents (DAAs) has revolutionized the treatment of hepatitis C. DAAs cure nearly all patients with short duration, oral treatments. Significant efforts are now underway to optimize DAA-based treatments. We discuss the potential role of interferon in this optimization. Clinical studies present compelling evidence that DAAs perform better in treatment-naive individuals than in individuals who previously failed treatment with interferon, a surprising correlation because interferon and DAAs are thought to act independently. Recent mathematical models explore a mechanistic hypothesis underlying this correlation. The hypothesis invokes the action of interferon at the cellular, individual, and population levels. Strong interferon responses prevent the productive infection of cells, reduce viral replication, and impede the development of resistance to DAAs in infected individuals and improve cure rates elicited by DAAs in treated populations. The models develop descriptions of these processes, integrate them into a comprehensive framework, and capture clinical data quantitatively, providing a successful test of the hypothesis. Individuals with strong endogenous interferon responses thus present a promising subpopulation for reducing DAA treatment durations. This review discusses the conceptual advances made by the models, highlights the new insights they unravel, and examines their applicability to optimize DAA-based treatments.
Keywords: bistability; direct-acting antiviral agents; interferon signaling; multiscale modeling; sustained virological response; viral kinetics.
© 2018 The Authors. Immunological Reviews Published by John Wiley & Sons Ltd.
Figures

Similar articles
-
Modelling how responsiveness to interferon improves interferon-free treatment of hepatitis C virus infection.PLoS Comput Biol. 2018 Jul 12;14(7):e1006335. doi: 10.1371/journal.pcbi.1006335. eCollection 2018 Jul. PLoS Comput Biol. 2018. PMID: 30001324 Free PMC article.
-
Understanding hepatitis C viral dynamics with direct-acting antiviral agents due to the interplay between intracellular replication and cellular infection dynamics.J Theor Biol. 2010 Dec 7;267(3):330-40. doi: 10.1016/j.jtbi.2010.08.036. Epub 2010 Sep 8. J Theor Biol. 2010. PMID: 20831874
-
Shift in disparities in hepatitis C treatment from interferon to DAA era: A population-based cohort study.J Viral Hepat. 2017 Aug;24(8):624-630. doi: 10.1111/jvh.12684. Epub 2017 Feb 20. J Viral Hepat. 2017. PMID: 28130810
-
Dasabuvir (ABT333) for the treatment of chronic HCV genotype I: a new face of cure, an expert review.Expert Rev Anti Infect Ther. 2016;14(2):157-65. doi: 10.1586/14787210.2016.1120668. Epub 2015 Dec 17. Expert Rev Anti Infect Ther. 2016. PMID: 26567871 Review.
-
Ribavirin revisited in the era of direct-acting antiviral therapy for hepatitis C virus infection.Liver Int. 2017 Jan;37(1):5-18. doi: 10.1111/liv.13212. Epub 2016 Sep 29. Liver Int. 2017. PMID: 27473533 Free PMC article. Review.
Cited by
-
Modeling recapitulates the heterogeneous outcomes of SARS-CoV-2 infection and quantifies the differences in the innate immune and CD8 T-cell responses between patients experiencing mild and severe symptoms.PLoS Pathog. 2022 Jun 27;18(6):e1010630. doi: 10.1371/journal.ppat.1010630. eCollection 2022 Jun. PLoS Pathog. 2022. PMID: 35759522 Free PMC article.
-
A dynamical motif comprising the interactions between antigens and CD8 T cells may underlie the outcomes of viral infections.Proc Natl Acad Sci U S A. 2019 Aug 27;116(35):17393-17398. doi: 10.1073/pnas.1902178116. Epub 2019 Aug 14. Proc Natl Acad Sci U S A. 2019. PMID: 31413198 Free PMC article.
-
Targeting TMPRSS2 and Cathepsin B/L together may be synergistic against SARS-CoV-2 infection.PLoS Comput Biol. 2020 Dec 8;16(12):e1008461. doi: 10.1371/journal.pcbi.1008461. eCollection 2020 Dec. PLoS Comput Biol. 2020. PMID: 33290397 Free PMC article.
-
Cost-Effectiveness Analysis of Pan-Genotypic Sofosbuvir-Based Regimens for Treatment of Chronic Hepatitis C Genotype 1 Infection in China.Front Public Health. 2021 Dec 9;9:779215. doi: 10.3389/fpubh.2021.779215. eCollection 2021. Front Public Health. 2021. PMID: 34957030 Free PMC article.
-
Immunomodulatory Role of Interferons in Viral and Bacterial Infections.Int J Mol Sci. 2023 Jun 14;24(12):10115. doi: 10.3390/ijms241210115. Int J Mol Sci. 2023. PMID: 37373262 Free PMC article. Review.
References
-
- Guidelines for the screening care and treatment of persons with chronic hepatitis C infection. Geneva: World Health Organization; 2016. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
