

Classification and management of adult inflammatory myopathies
- PMID: 30129477
- PMCID: PMC11646336
- DOI: 10.1016/S1474-4422(18)30254-0
Classification and management of adult inflammatory myopathies
Abstract
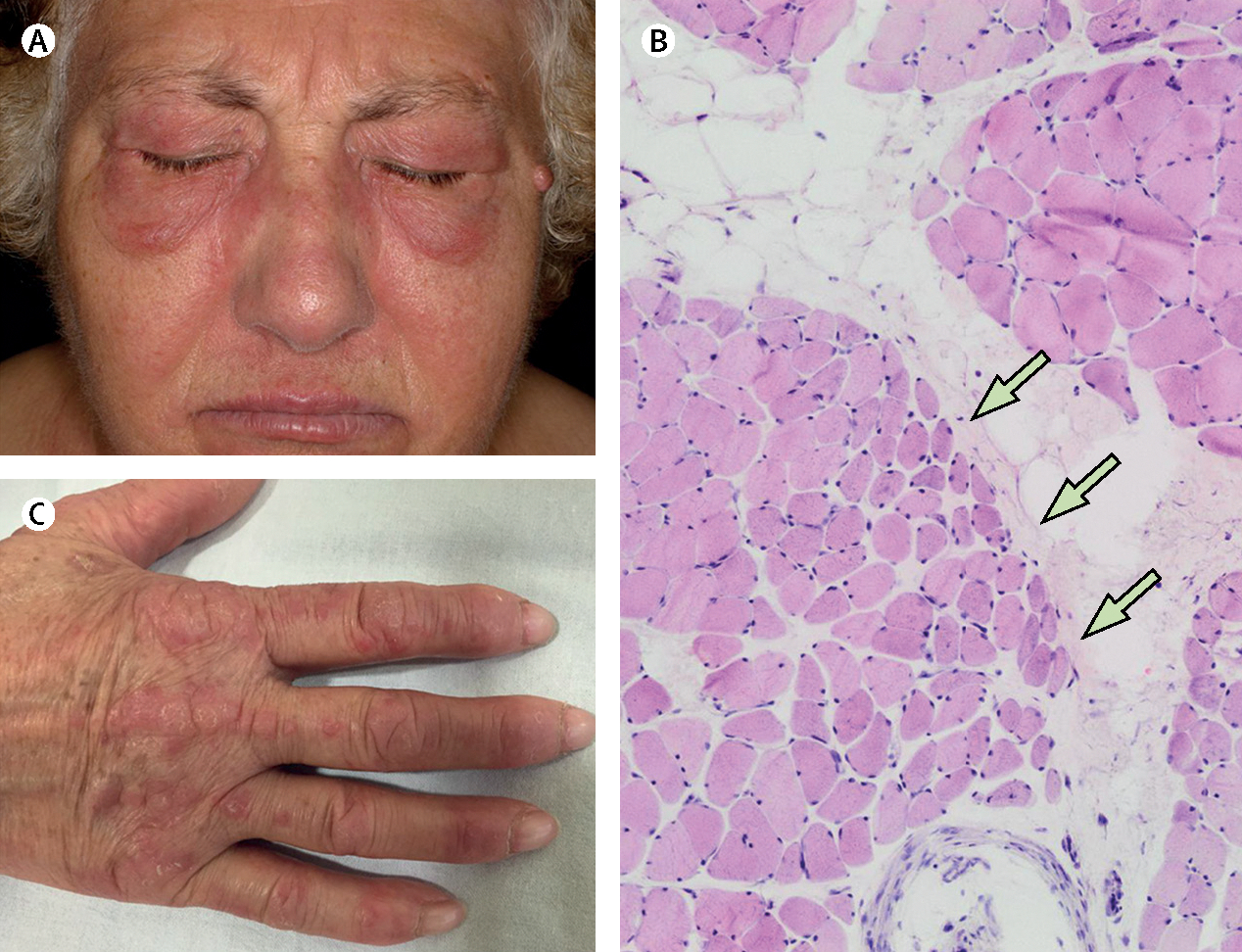
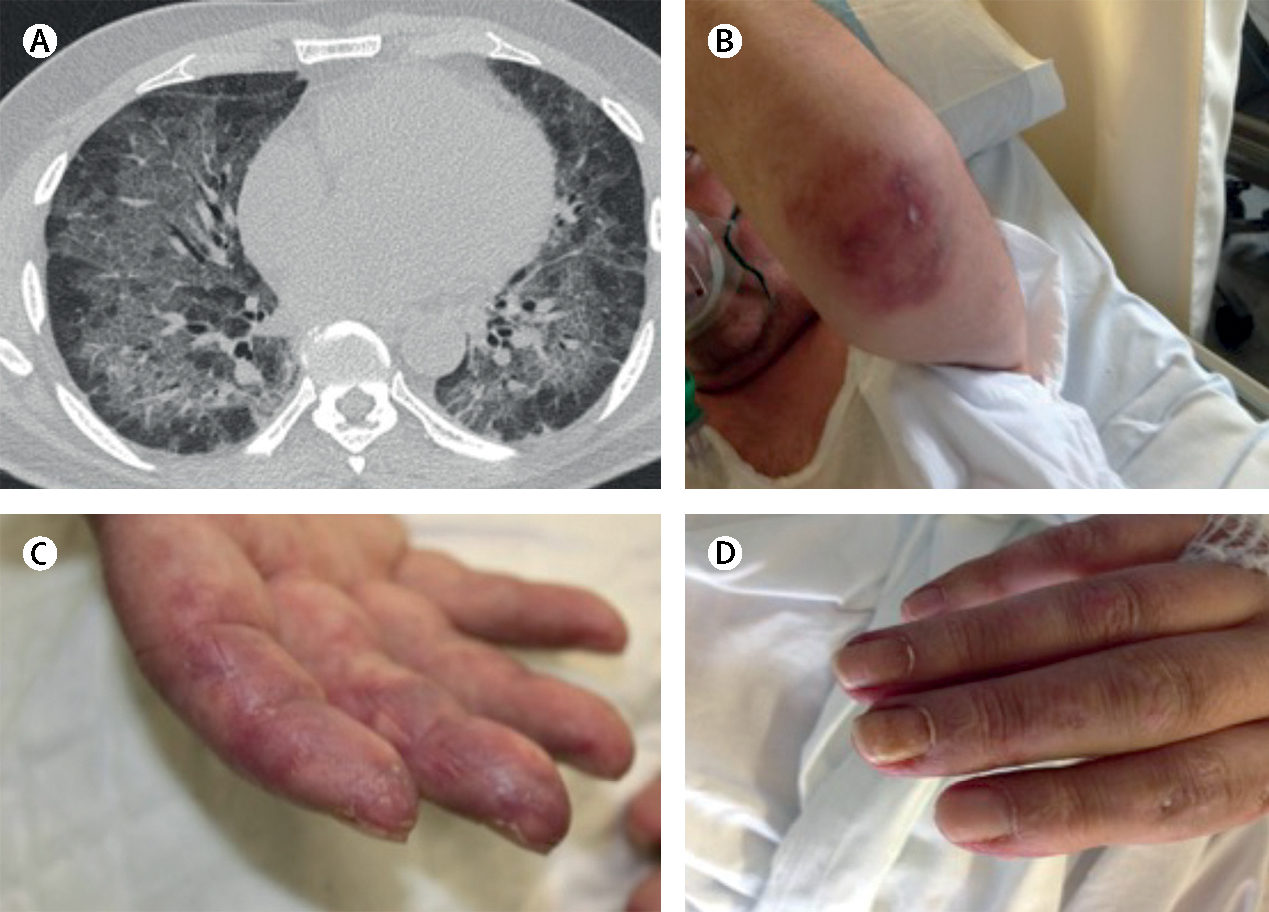
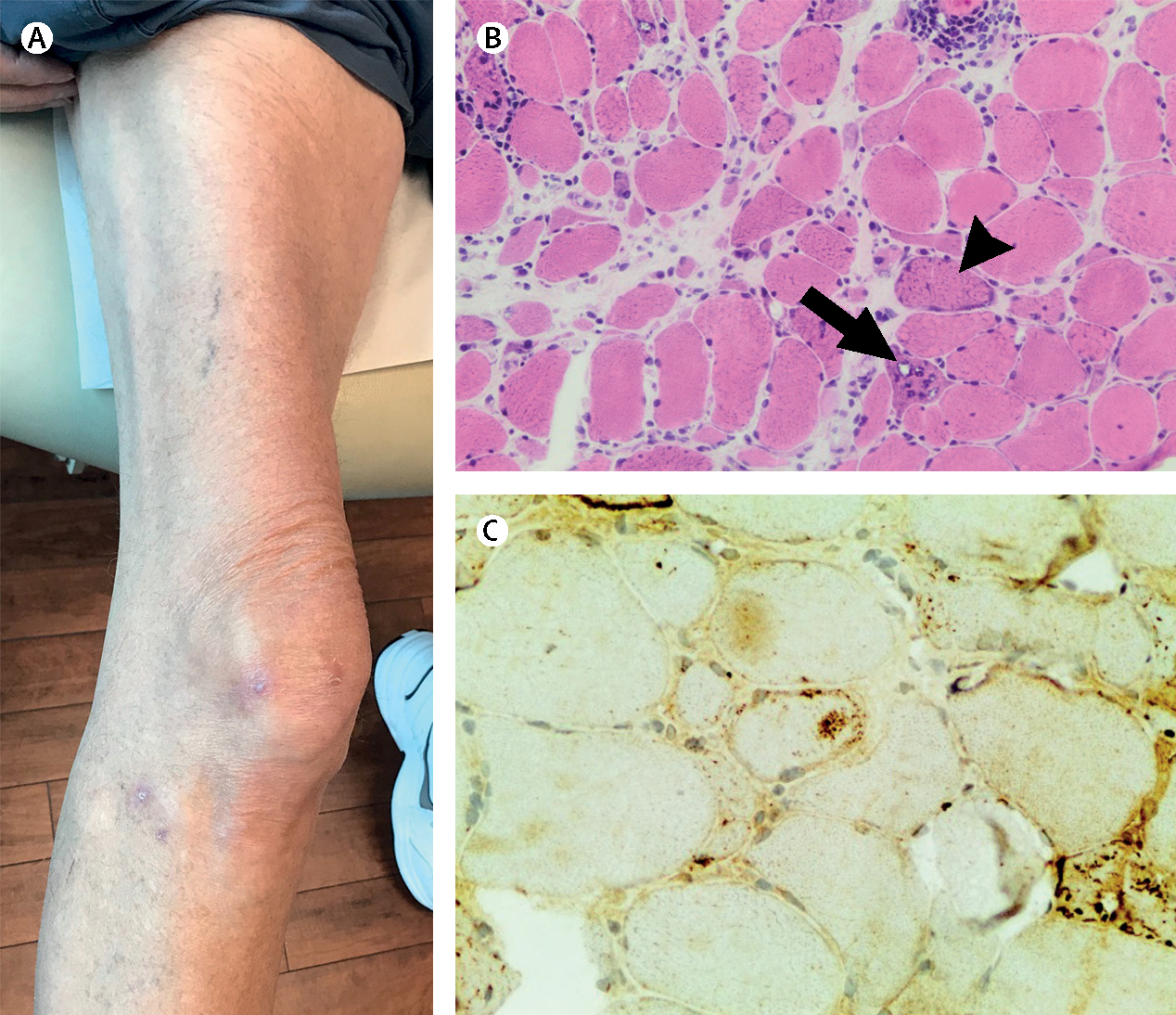
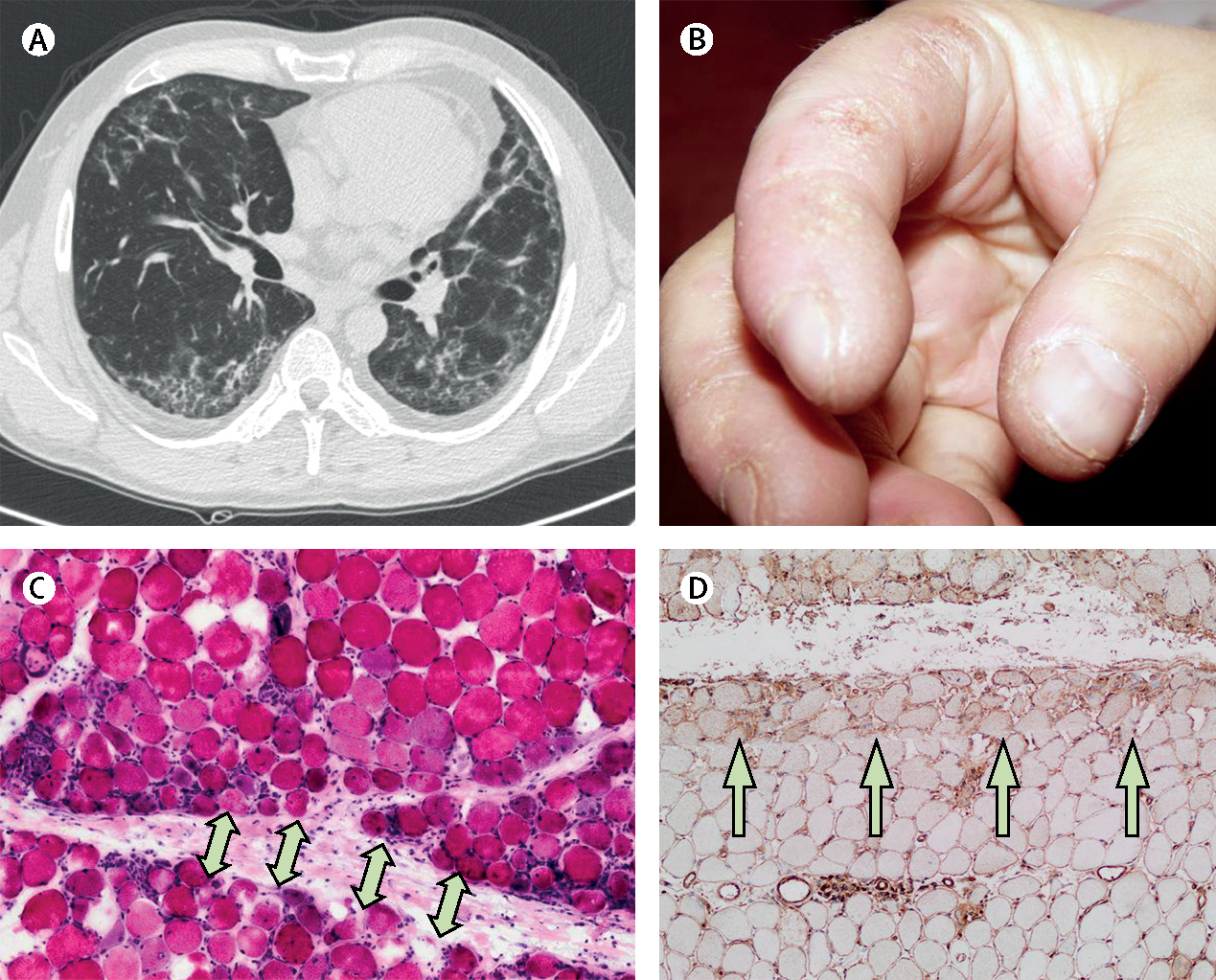
Inflammatory myopathies, collectively known as myositis, are heterogeneous disorders characterised by muscle inflammation, and frequently accompanied by extramuscular manifestations that affect the skin, lung, and joints. Patients with inflammatory myopathies were previously classified as having dermatomyositis if characteristic rashes accompanied the muscle involvement, and as having polymyositis if no rashes were present. Five main types of inflammatory myopathies are now widely recognised: dermatomyositis, immune-mediated necrotising myopathy, sporadic inclusion-body myositis, overlap myositis (including antisynthetase syndrome), and polymyositis. The discovery of autoantibodies that are specifically associated with characteristic clinical phenotypes has been instrumental to the understanding of inflammatory myopathies. Treatment is still largely based on expert opinion, but several studies have shown effectiveness of different therapies in various subsets of inflammatory myopathies. These advances will undoubtedly improve the outcomes of patients with inflammatory myopathies.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests
ALM has a patent on an anti-HMGCR assay. AS-O’C, IP-F, ET-A, JCM, and JMG-J declare no competing interests.
Figures
References
-
- Senecal JL, Raynauld JP, Troyanov Y. Editorial: a new classification of adult autoimmune myositis. Arthritis Rheumatol 2017; 69: 878–84. - PubMed
-
- Dalakas MC. Inflammatory muscle diseases. N Engl J Med 2015; 373: 393–94. - PubMed
-
- Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med 2016; 280: 8–23. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
