

Iron and Cancer
- PMID: 30130469
- PMCID: PMC8118195
- DOI: 10.1146/annurev-nutr-082117-051732
Iron and Cancer
Abstract
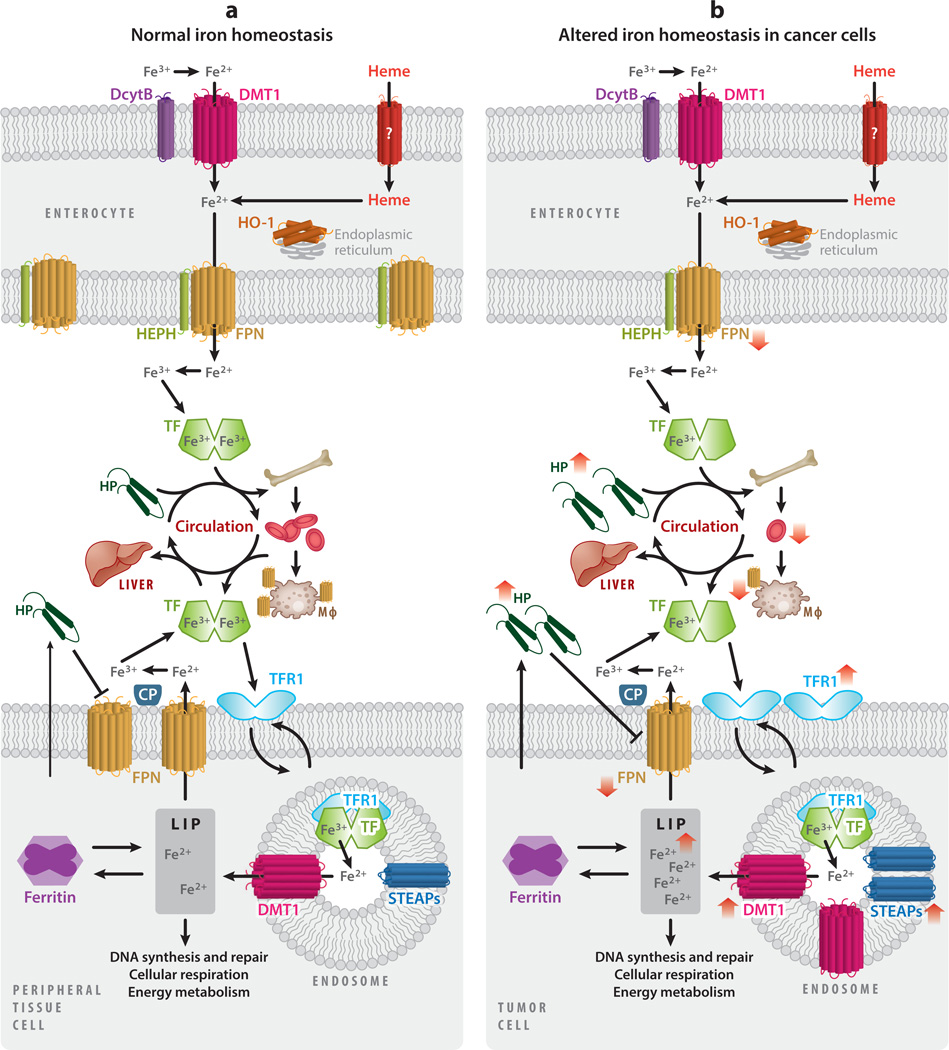
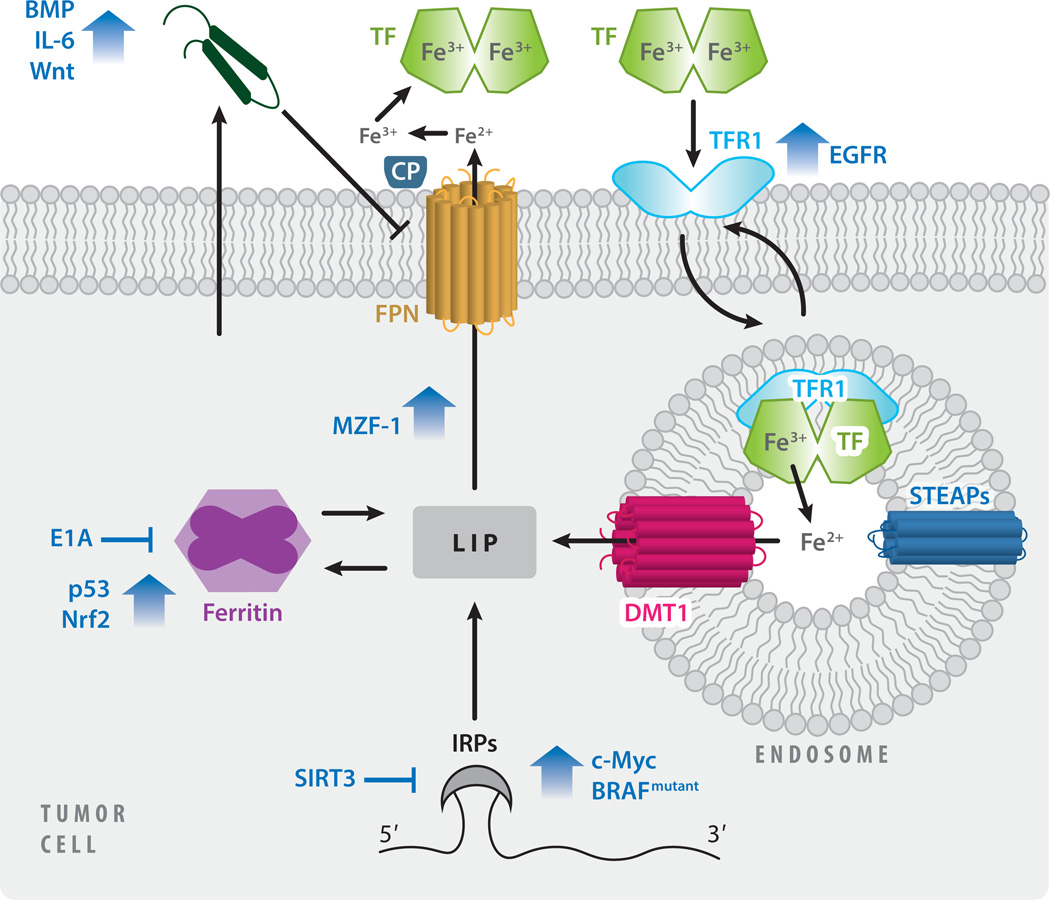
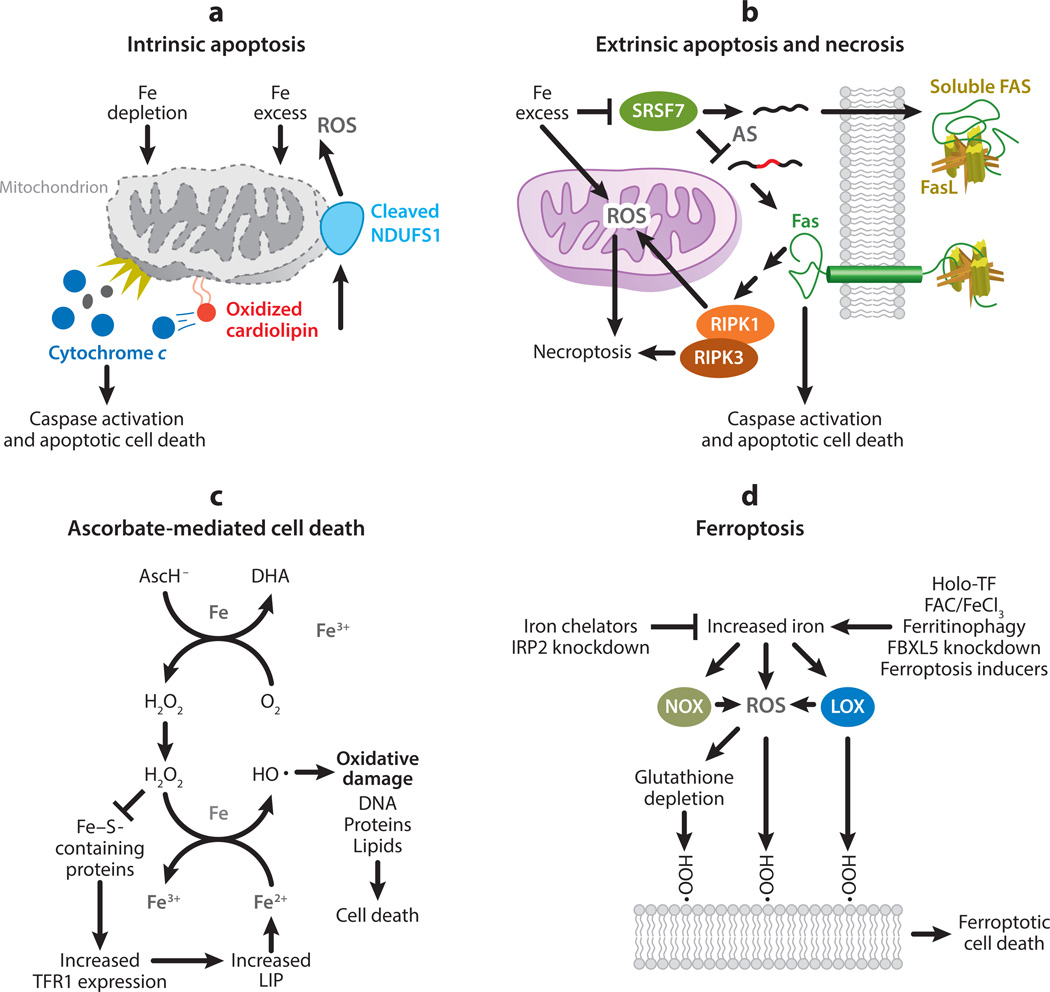
This review explores the multifaceted role that iron has in cancer biology. Epidemiological studies have demonstrated an association between excess iron and increased cancer incidence and risk, while experimental studies have implicated iron in cancer initiation, tumor growth, and metastasis. The roles of iron in proliferation, metabolism, and metastasis underpin the association of iron with tumor growth and progression. Cancer cells exhibit an iron-seeking phenotype achieved through dysregulation of iron metabolic proteins. These changes are mediated, at least in part, by oncogenes and tumor suppressors. The dependence of cancer cells on iron has implications in a number of cell death pathways, including ferroptosis, an iron-dependent form of cell death. Uniquely, both iron excess and iron depletion can be utilized in anticancer therapies. Investigating the efficacy of these therapeutic approaches is an area of active research that promises substantial clinical impact.
Keywords: cancer; carcinogenesis; cell death; iron.
Figures
References
-
- Abeysinghe RD, Greene BT, Haynes R, Willingham MC, Turner J, et al. 2001. p53-independent apoptosis mediated by tachpyridine, an anti-cancer iron chelator. Carcinogenesis 22:1607–14 - PubMed
-
- Adams PC. 2015. Epidemiology and diagnostic testing for hemochromatosis and iron overload. Int. J. Lab. Hematol. 37(Suppl. 1):25–30 - PubMed
-
- Aisen P, Enns C, Wessling-Resnick M. 2001. Chemistry and biology of eukaryotic iron metabolism. Int. J. Biochem. Cell Biol. 33:940–59 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
