

MARVEL, a Tool for Prediction of Bacteriophage Sequences in Metagenomic Bins
- PMID: 30131825
- PMCID: PMC6090037
- DOI: 10.3389/fgene.2018.00304
MARVEL, a Tool for Prediction of Bacteriophage Sequences in Metagenomic Bins
Abstract
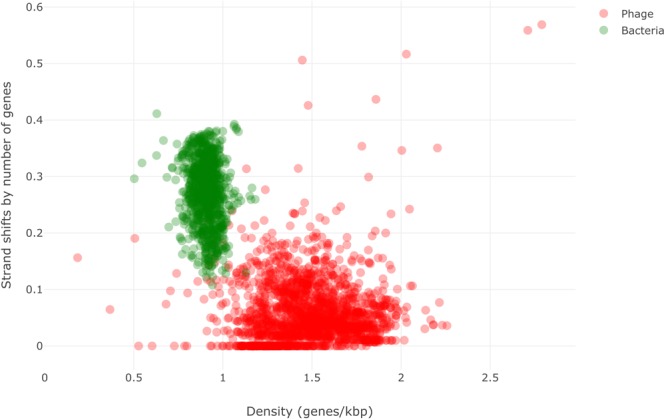
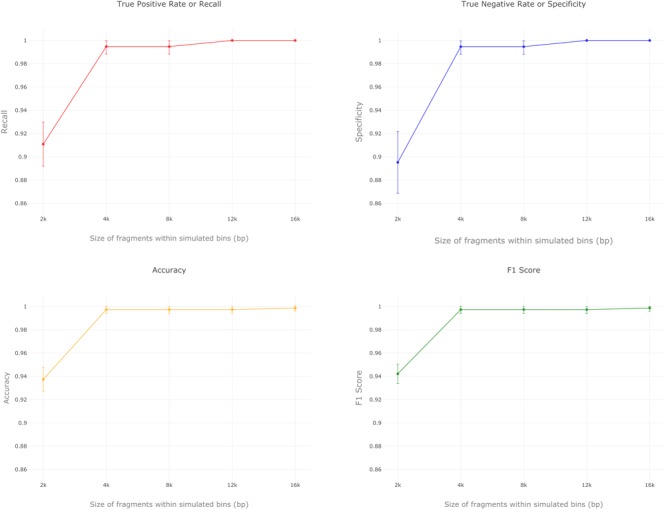
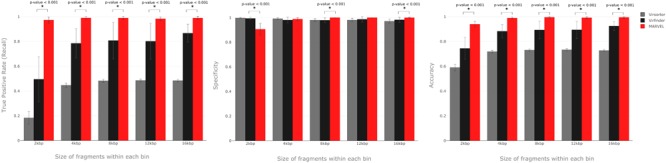
Here we present MARVEL, a tool for prediction of double-stranded DNA bacteriophage sequences in metagenomic bins. MARVEL uses a random forest machine learning approach. We trained the program on a dataset with 1,247 phage and 1,029 bacterial genomes, and tested it on a dataset with 335 bacterial and 177 phage genomes. We show that three simple genomic features extracted from contig sequences were sufficient to achieve a good performance in separating bacterial from phage sequences: gene density, strand shifts, and fraction of significant hits to a viral protein database. We compared the performance of MARVEL to that of VirSorter and VirFinder, two popular programs for predicting viral sequences. Our results show that all three programs have comparable specificity, but MARVEL achieves much better performance on the recall (sensitivity) measure. This means that MARVEL should be able to identify many more phage sequences in metagenomic bins than heretofore has been possible. In a simple test with real data, containing mostly bacterial sequences, MARVEL classified 58 out of 209 bins as phage genomes; other evidence suggests that 57 of these 58 bins are novel phage sequences. MARVEL is freely available at https://github.com/LaboratorioBioinformatica/MARVEL.
Keywords: machine learning; microbiome; phage; random forest; virus.
Figures
References
-
- Amgarten D., Martins L. F., Lombardi K. C., Antunes L. P., de Souza A. P. S., Nicastro G. G., et al. (2017). Three novel Pseudomonas phages isolated from composting provide insights into the evolution and diversity of tailed phages. BMC Genomics 18:346. 10.1186/s12864-017-3729-z - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources