

The TAB1-p38α complex aggravates myocardial injury and can be targeted by small molecules
- PMID: 30135318
- PMCID: PMC6141180
- DOI: 10.1172/jci.insight.121144
The TAB1-p38α complex aggravates myocardial injury and can be targeted by small molecules
Abstract
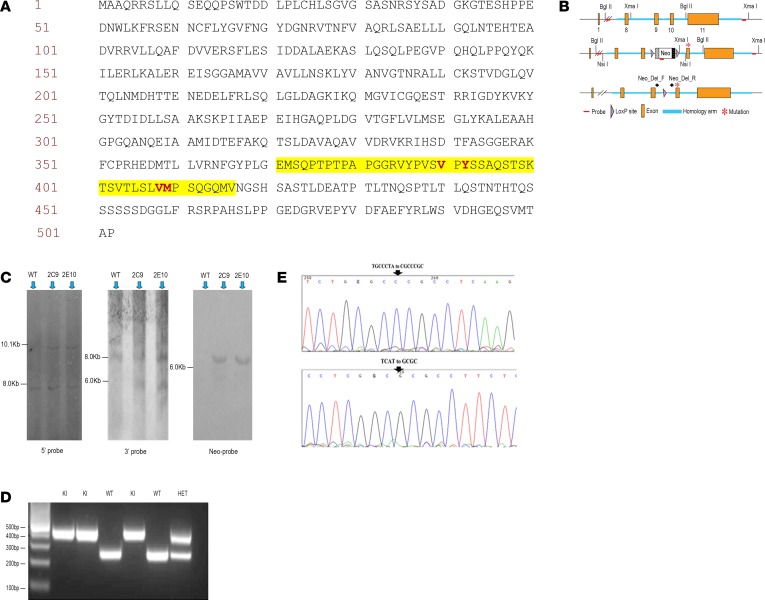
Inhibiting MAPK14 (p38α) diminishes cardiac damage in myocardial ischemia. During myocardial ischemia, p38α interacts with TAB1, a scaffold protein, which promotes p38α autoactivation; active p38α (pp38α) then transphosphorylates TAB1. Previously, we solved the X-ray structure of the p38α-TAB1 (residues 384-412) complex. Here, we further characterize the interaction by solving the structure of the pp38α-TAB1 (residues 1-438) complex in the active state. Based on this information, we created a global knock-in (KI) mouse with substitution of 4 residues on TAB1 that we show are required for docking onto p38α. Whereas ablating p38α or TAB1 resulted in early embryonal lethality, the TAB1-KI mice were viable and had no appreciable alteration in their lymphocyte repertoire or myocardial transcriptional profile; nonetheless, following in vivo regional myocardial ischemia, infarction volume was significantly reduced and the transphosphorylation of TAB1 was disabled. Unexpectedly, the activation of myocardial p38α during ischemia was only mildly attenuated in TAB1-KI hearts. We also identified a group of fragments able to disrupt the interaction between p38α and TAB1. We conclude that the interaction between the 2 proteins can be targeted with small molecules. The data reveal that it is possible to selectively inhibit signaling downstream of p38α to attenuate ischemic injury.
Keywords: Cardiology; Pharmacology; Protein kinases; Structural biology; Therapeutics.
Conflict of interest statement
Figures









Similar articles
-
TAB1-Induced Autoactivation of p38α Mitogen-Activated Protein Kinase Is Crucially Dependent on Threonine 185.Mol Cell Biol. 2018 Feb 12;38(5):e00409-17. doi: 10.1128/MCB.00409-17. Print 2018 Mar 1. Mol Cell Biol. 2018. PMID: 29229647 Free PMC article.
-
Determinants that control the specific interactions between TAB1 and p38alpha.Mol Cell Biol. 2006 May;26(10):3824-34. doi: 10.1128/MCB.26.10.3824-3834.2006. Mol Cell Biol. 2006. PMID: 16648477 Free PMC article.
-
Disruption of TAB1/p38α interaction using a cell-permeable peptide limits myocardial ischemia/reperfusion injury.Mol Ther. 2013 Sep;21(9):1668-77. doi: 10.1038/mt.2013.90. Epub 2013 Jul 23. Mol Ther. 2013. PMID: 23877036 Free PMC article.
-
Mechanism and consequence of the autoactivation of p38α mitogen-activated protein kinase promoted by TAB1.Nat Struct Mol Biol. 2013 Oct;20(10):1182-90. doi: 10.1038/nsmb.2668. Epub 2013 Sep 15. Nat Struct Mol Biol. 2013. PMID: 24037507 Free PMC article.
-
Diverse mechanisms of myocardial p38 mitogen-activated protein kinase activation: evidence for MKK-independent activation by a TAB1-associated mechanism contributing to injury during myocardial ischemia.Circ Res. 2003 Aug 8;93(3):254-61. doi: 10.1161/01.RES.0000083490.43943.85. Epub 2003 Jun 26. Circ Res. 2003. PMID: 12829618
Cited by
-
Fluorescence resonance energy transfer (FRET) spatiotemporal mapping of atypical P38 reveals an endosomal and cytosolic spatial bias.Sci Rep. 2023 May 8;13(1):7477. doi: 10.1038/s41598-023-33953-y. Sci Rep. 2023. PMID: 37156828 Free PMC article.
-
Spatiotemporal control of kinases and the biomolecular tools to trace activity.J Biol Chem. 2024 Nov;300(11):107846. doi: 10.1016/j.jbc.2024.107846. Epub 2024 Oct 1. J Biol Chem. 2024. PMID: 39362469 Free PMC article. Review.
-
Atypical p38 Signaling, Activation, and Implications for Disease.Int J Mol Sci. 2021 Apr 17;22(8):4183. doi: 10.3390/ijms22084183. Int J Mol Sci. 2021. PMID: 33920735 Free PMC article. Review.
-
Ubiquitination as a Key Regulator of Endosomal Signaling by GPCRs.Front Cell Dev Biol. 2019 Mar 29;7:43. doi: 10.3389/fcell.2019.00043. eCollection 2019. Front Cell Dev Biol. 2019. PMID: 30984758 Free PMC article. Review.
-
Subcellular hot spots of GPCR signaling promote vascular inflammation.Curr Opin Endocr Metab Res. 2021 Feb;16:37-42. doi: 10.1016/j.coemr.2020.07.011. Epub 2020 Aug 18. Curr Opin Endocr Metab Res. 2021. PMID: 32838054 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
- RG/12/12/29872/BHF_/British Heart Foundation/United Kingdom
- MC_PC_17164/MRC_/Medical Research Council/United Kingdom
- MR/K003232/1/MRC_/Medical Research Council/United Kingdom
- BB/C503646/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- RG/17/16/33294/BHF_/British Heart Foundation/United Kingdom
- DH_/Department of Health/United Kingdom
- PG/15/26/31373/BHF_/British Heart Foundation/United Kingdom
- G0600785/MRC_/Medical Research Council/United Kingdom
- MR/R01065X/1/MRC_/Medical Research Council/United Kingdom
- G1000458/MRC_/Medical Research Council/United Kingdom
- FS/11/45/28859/BHF_/British Heart Foundation/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- PG/13/13/30018/BHF_/British Heart Foundation/United Kingdom
- FS/14/29/30896/BHF_/British Heart Foundation/United Kingdom
- MR/J007501/1/MRC_/Medical Research Council/United Kingdom
- G0700320/MRC_/Medical Research Council/United Kingdom
- MR/L009684/1/MRC_/Medical Research Council/United Kingdom
- PG/10/98/28655/BHF_/British Heart Foundation/United Kingdom
- PG/17/44/33064/BHF_/British Heart Foundation/United Kingdom
- MR/P023150/1/MRC_/Medical Research Council/United Kingdom
- SP/14/2/30922/BHF_/British Heart Foundation/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
