

Genomic patterns of progression in smoldering multiple myeloma
- PMID: 30135448
- PMCID: PMC6105687
- DOI: 10.1038/s41467-018-05058-y
Genomic patterns of progression in smoldering multiple myeloma
Abstract
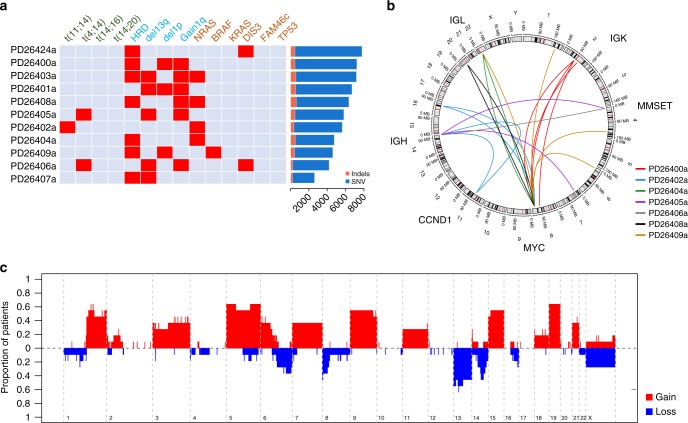
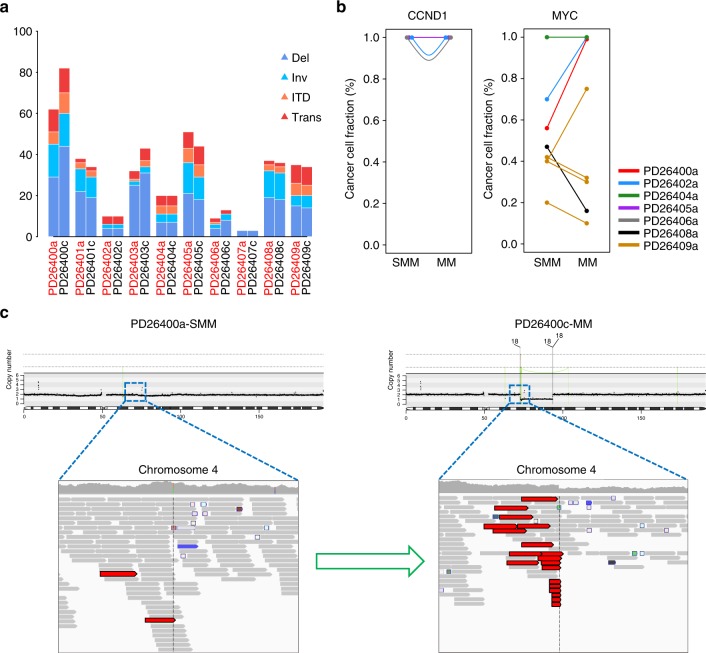
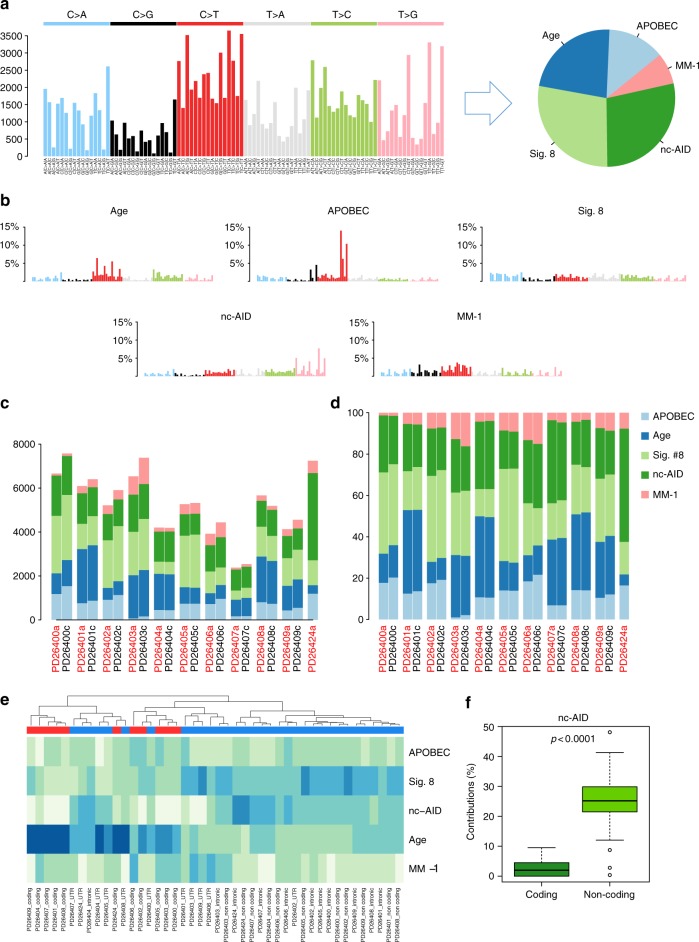
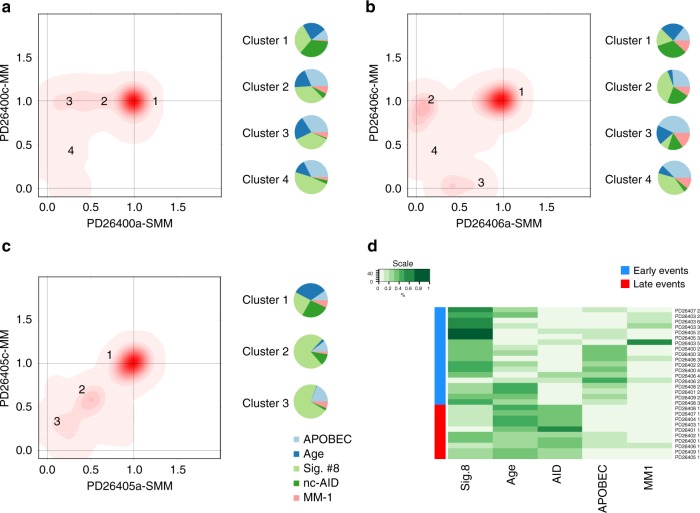
We analyzed whole genomes of unique paired samples from smoldering multiple myeloma (SMM) patients progressing to multiple myeloma (MM). We report that the genomic landscape, including mutational profile and structural rearrangements at the smoldering stage is very similar to MM. Paired sample analysis shows two different patterns of progression: a "static progression model", where the subclonal architecture is retained as the disease progressed to MM suggesting that progression solely reflects the time needed to accumulate a sufficient disease burden; and a "spontaneous evolution model", where a change in the subclonal composition is observed. We also observe that activation-induced cytidine deaminase plays a major role in shaping the mutational landscape of early subclinical phases, while progression is driven by APOBEC cytidine deaminases. These results provide a unique insight into myelomagenesis with potential implications for the definition of smoldering disease and timing of treatment initiation.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Manier, S. et al. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol.14, 100–113 (2017). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
