

Glycosylation Significantly Inhibits the Aggregation of Human Prion Protein and Decreases Its Cytotoxicity
- PMID: 30135544
- PMCID: PMC6105643
- DOI: 10.1038/s41598-018-30770-6
Glycosylation Significantly Inhibits the Aggregation of Human Prion Protein and Decreases Its Cytotoxicity
Erratum in
-
Author Correction: Glycosylation Significantly Inhibits the Aggregation of Human Prion Protein and Decreases Its Cytotoxicity.Sci Rep. 2018 Sep 4;8(1):13486. doi: 10.1038/s41598-018-31650-9. Sci Rep. 2018. PMID: 30177728 Free PMC article.
Abstract
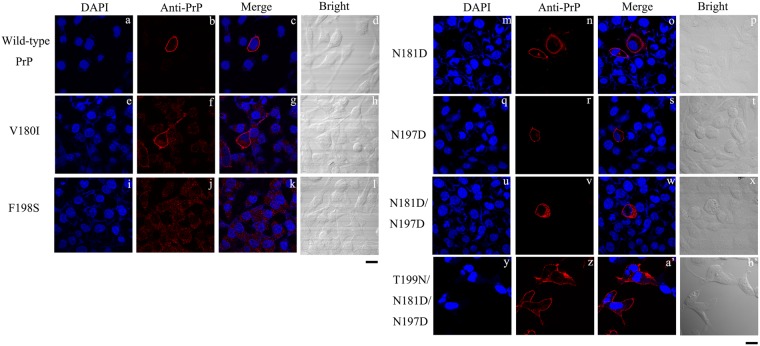
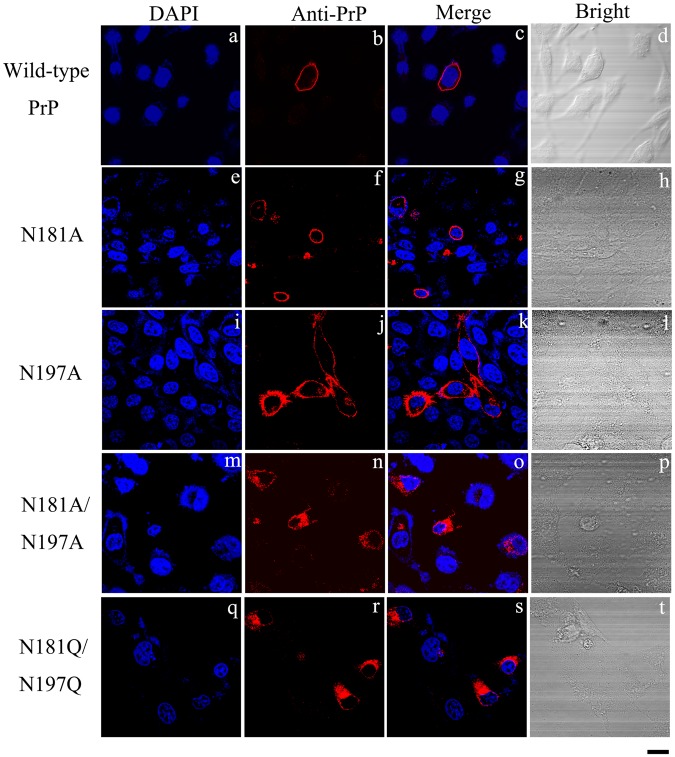
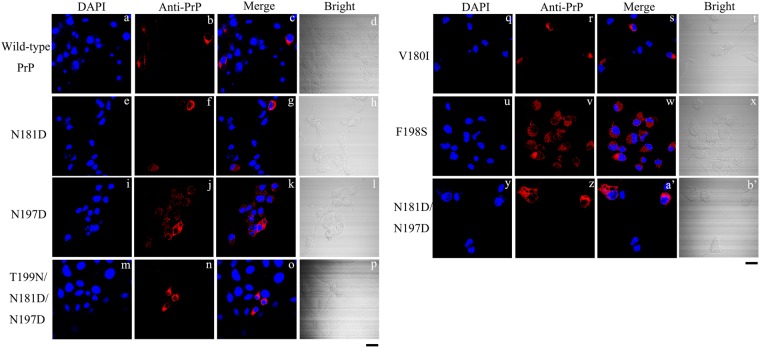
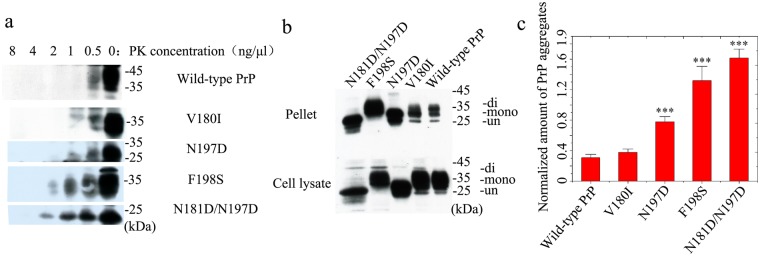
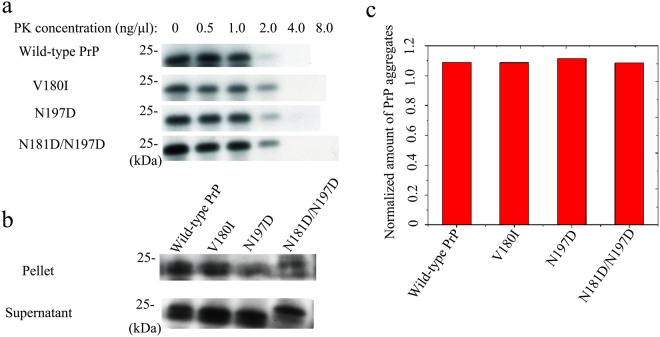
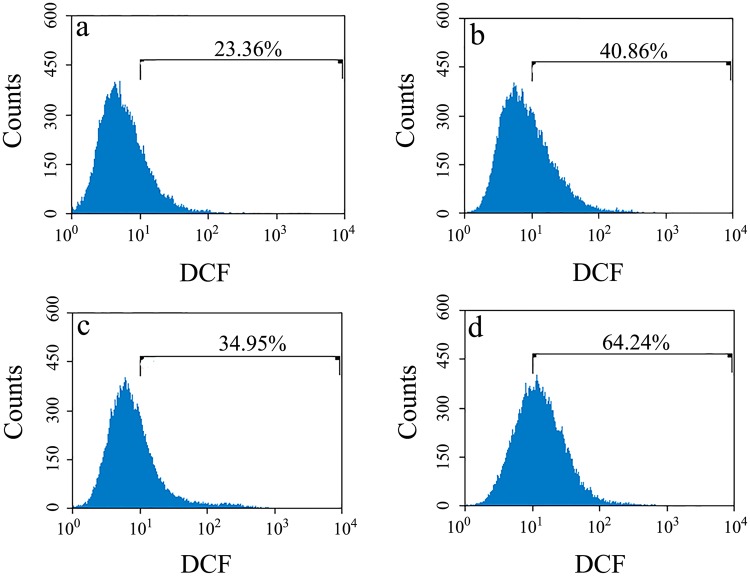
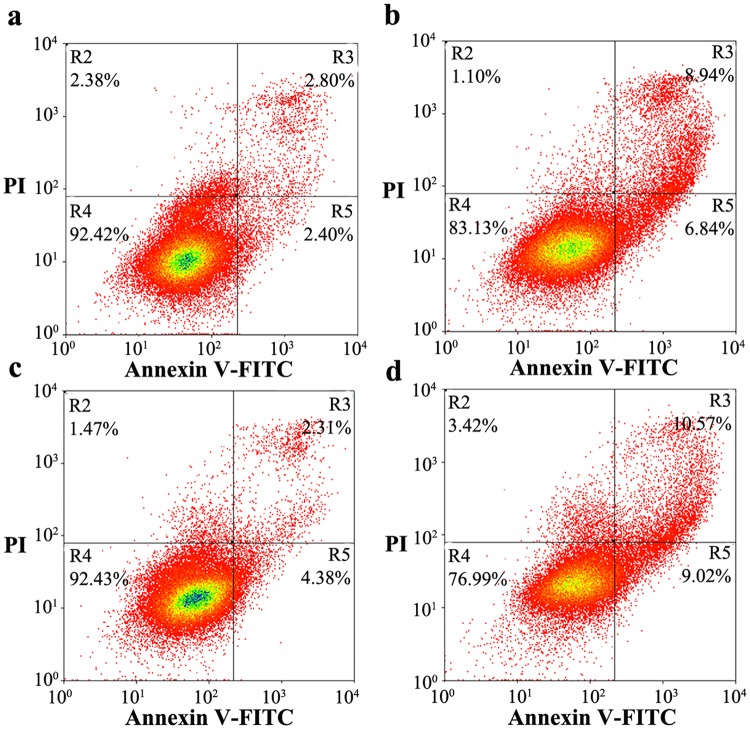
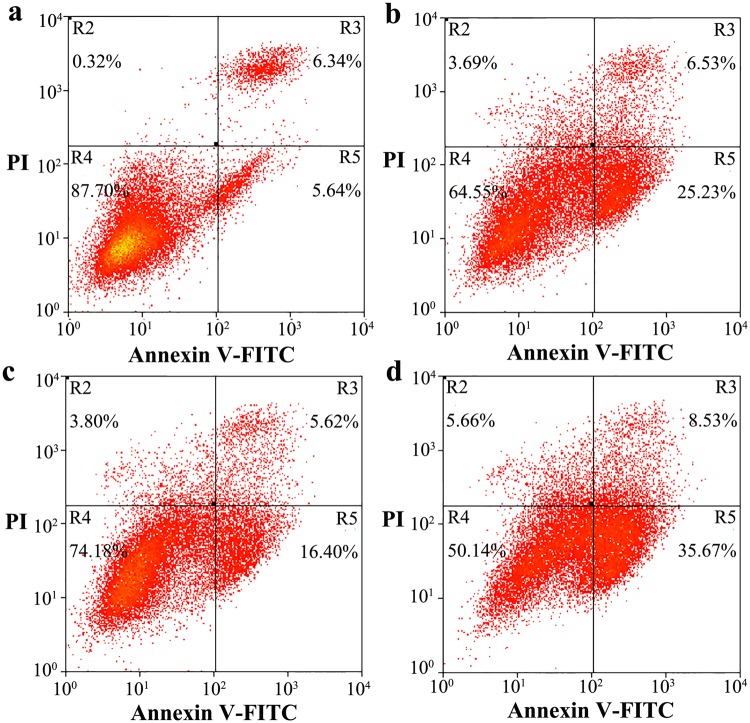
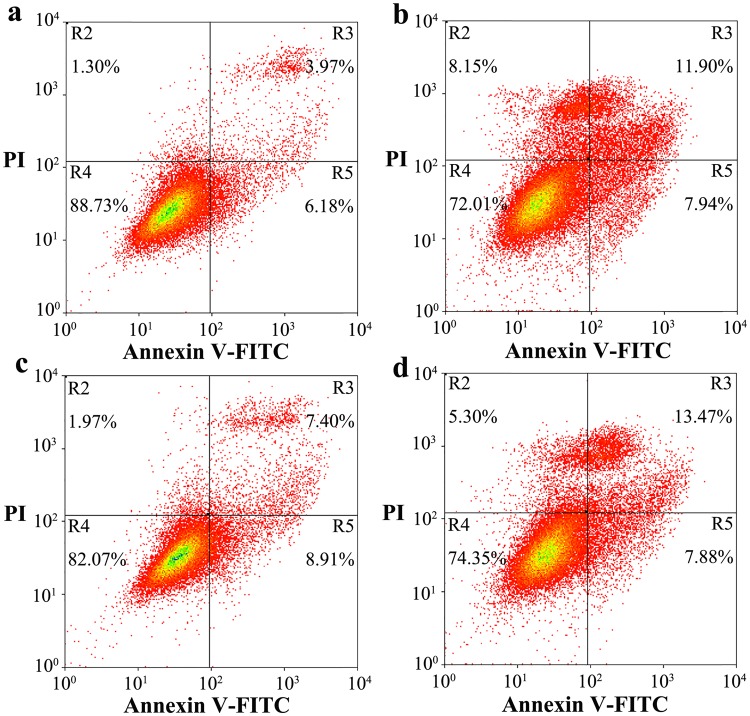
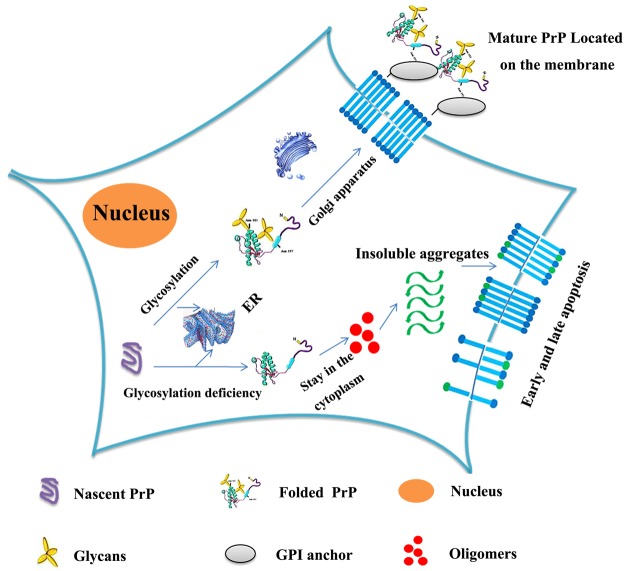
Prion diseases are primarily caused by the misfolding of prion proteins in humans, cattle, sheep, and cervid species. The effects of glycosylation on prion protein (PrP) structure and function have not been thoroughly elucidated to date. In this study, we attempt to elucidate the effects of glycosylation on the aggregation and toxicity of human PrP. As revealed by immunocytochemical staining, wild-type PrP and its monoglycosylated mutants N181D, N197D, and T199N/N181D/N197D are primarily attached to the plasma membrane. In contrast, PrP F198S, a pathological mutant with an altered residue within the glycosylation site, and an unglycosylated PrP mutant, N181D/N197D, primarily exist in the cytoplasm. In the pathological mutant V180I, there is an equal mix of membranous and cytoplasmic PrP, indicating that N-linked glycosylation deficiency impairs the correct localization of human PrP at the plasma membrane. As shown by immunoblotting and flow cytometry, human PrP located in the cytoplasm displays considerably greater PK resistance and aggregation ability and is associated with considerably higher cellular ROS levels than PrP located on the plasma membrane. Furthermore, glycosylation deficiency enhances human PrP cytotoxicity induced by MG132 or the toxic prion peptide PrP 106-126. Therefore, we propose that glycosylation acts as a necessary cofactor in determining PrP localization on the plasma membrane and that it significantly inhibits the aggregation of human PrP and decreases its cytotoxicity.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
