

Allergic inflammatory memory in human respiratory epithelial progenitor cells
- PMID: 30135581
- PMCID: PMC6133715
- DOI: 10.1038/s41586-018-0449-8
Allergic inflammatory memory in human respiratory epithelial progenitor cells
Abstract
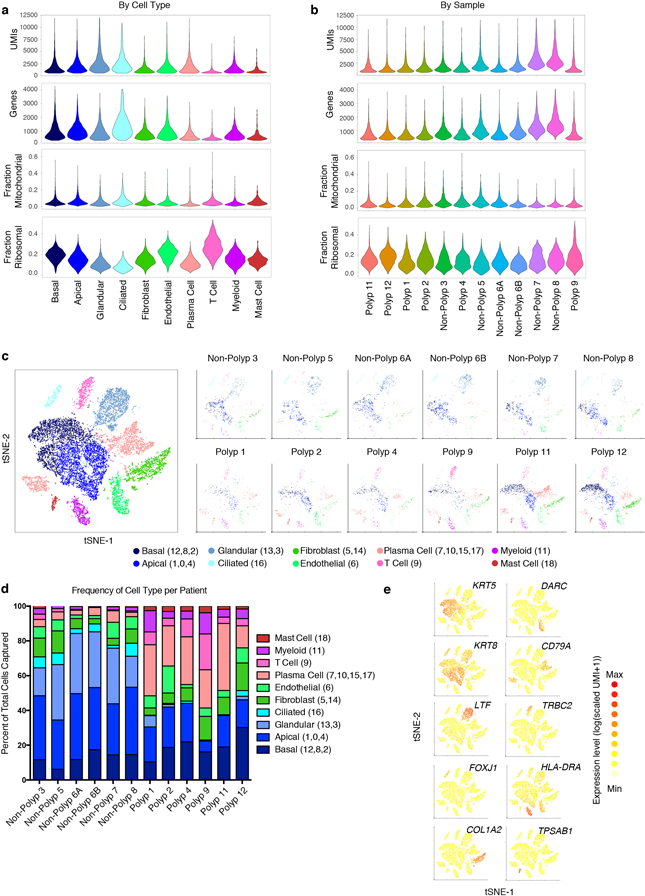
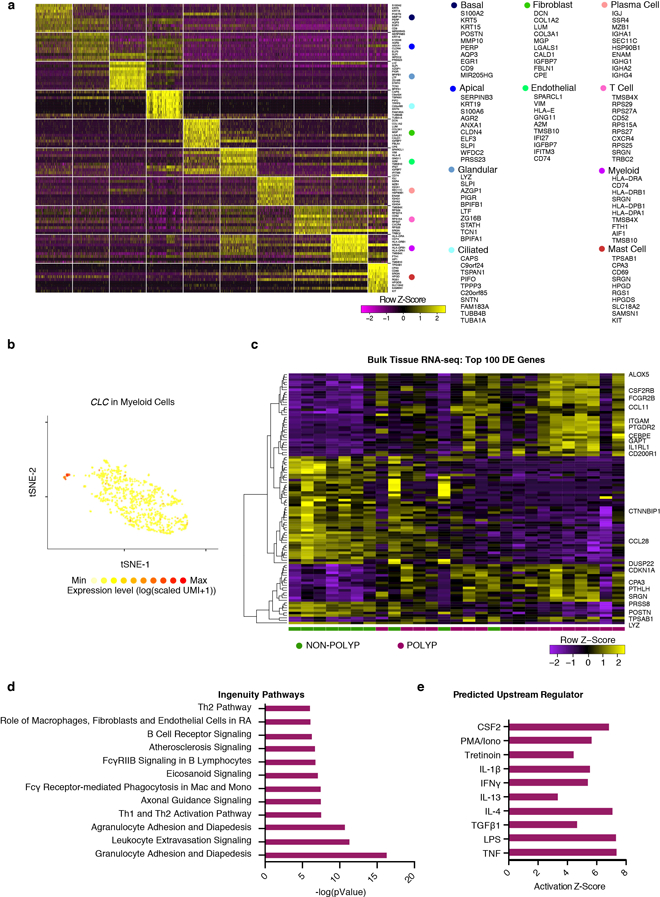
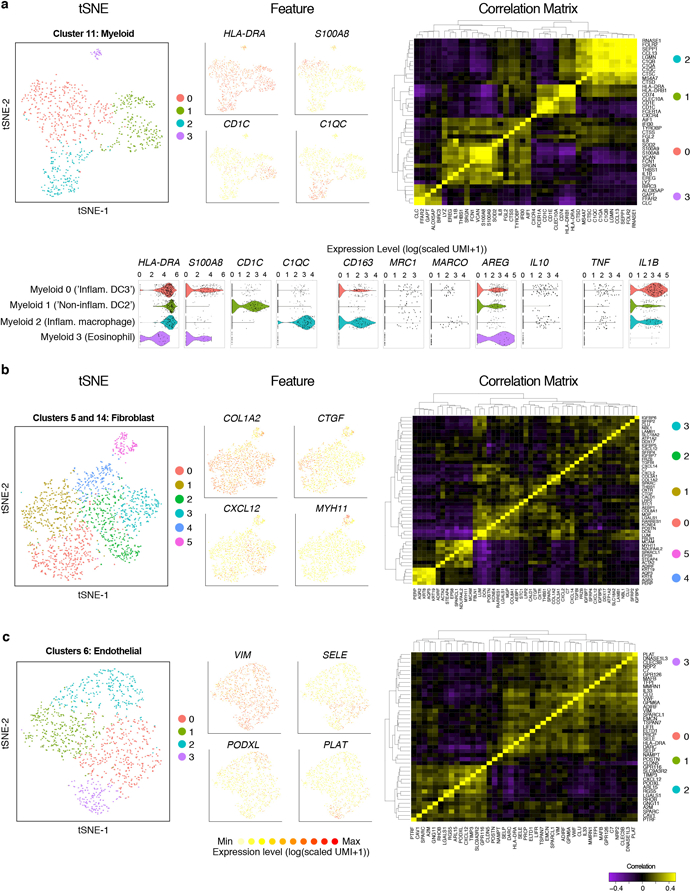
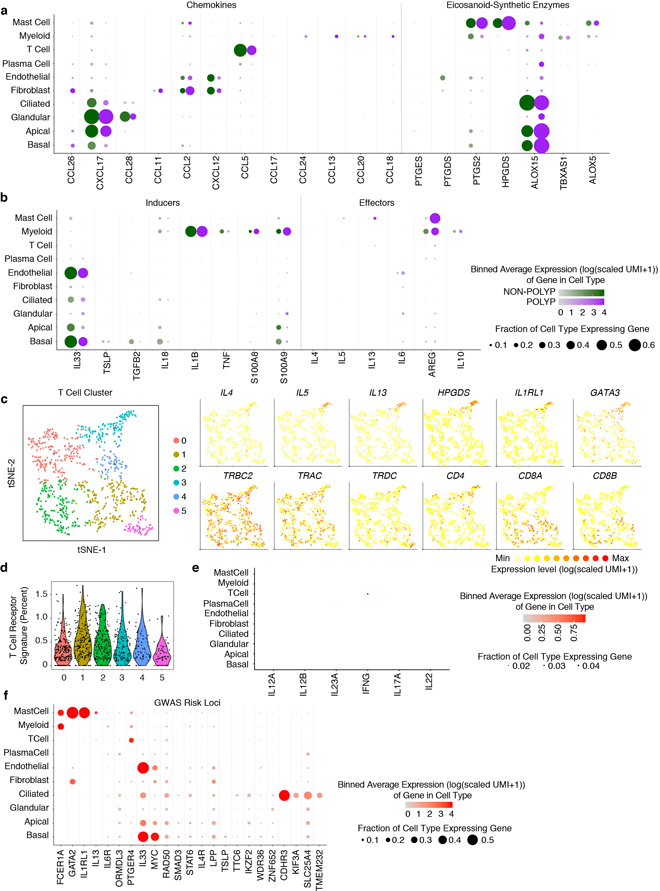
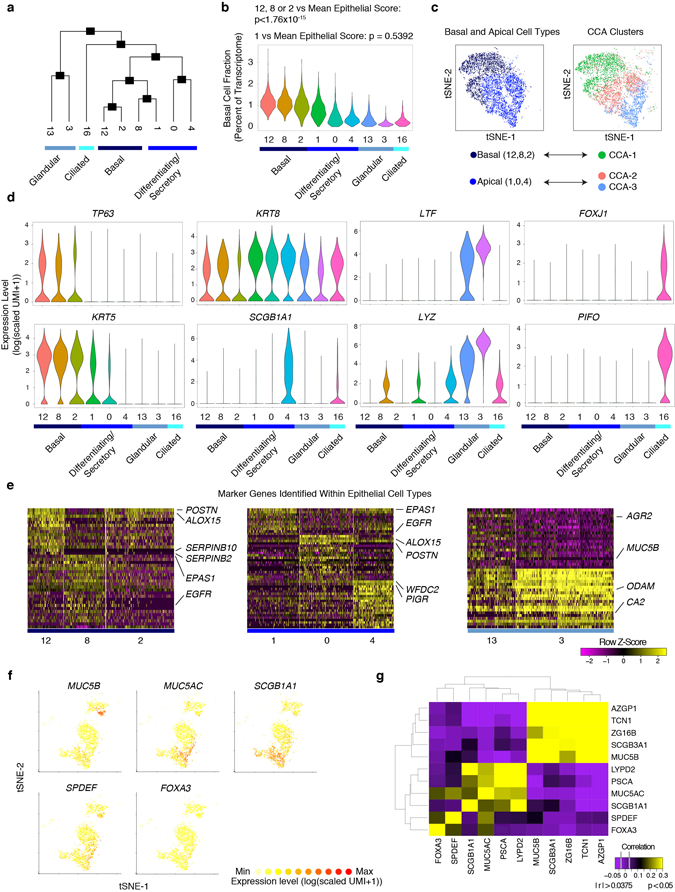
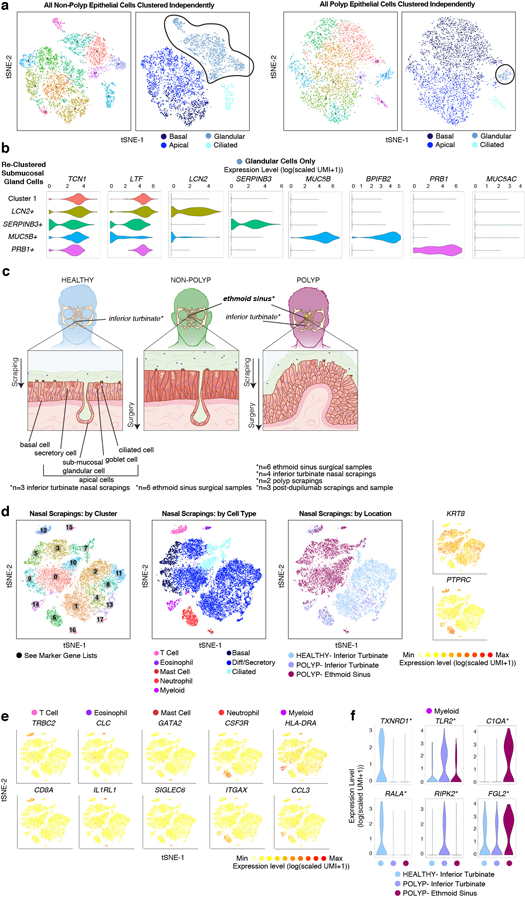
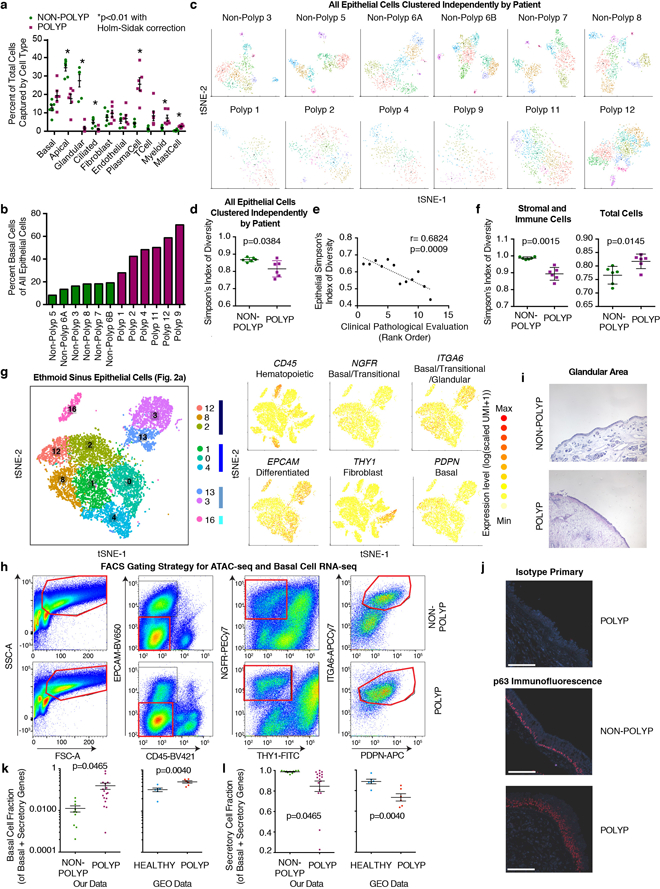
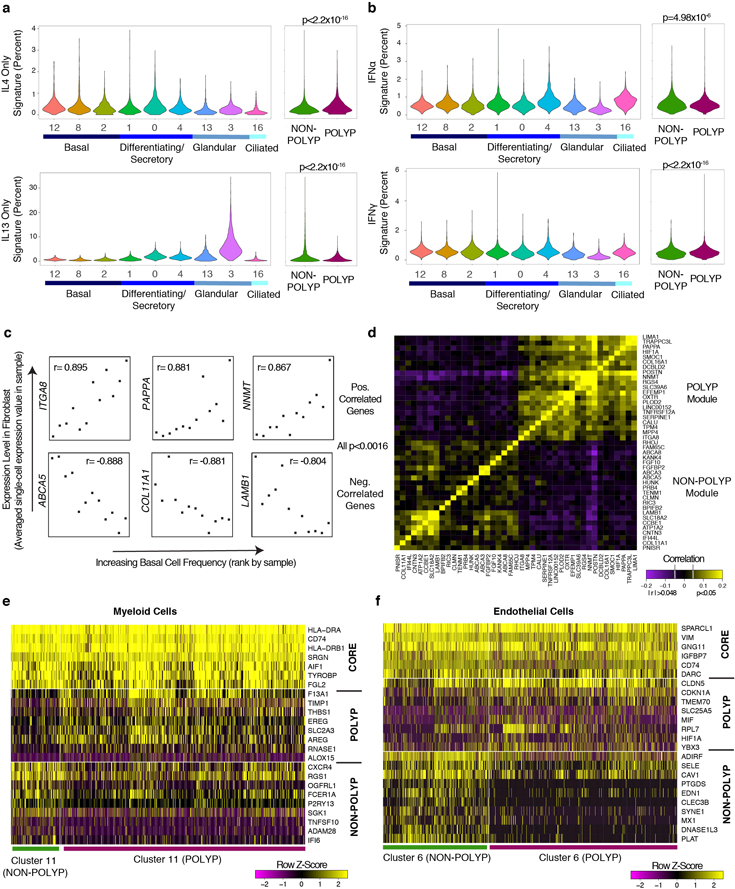
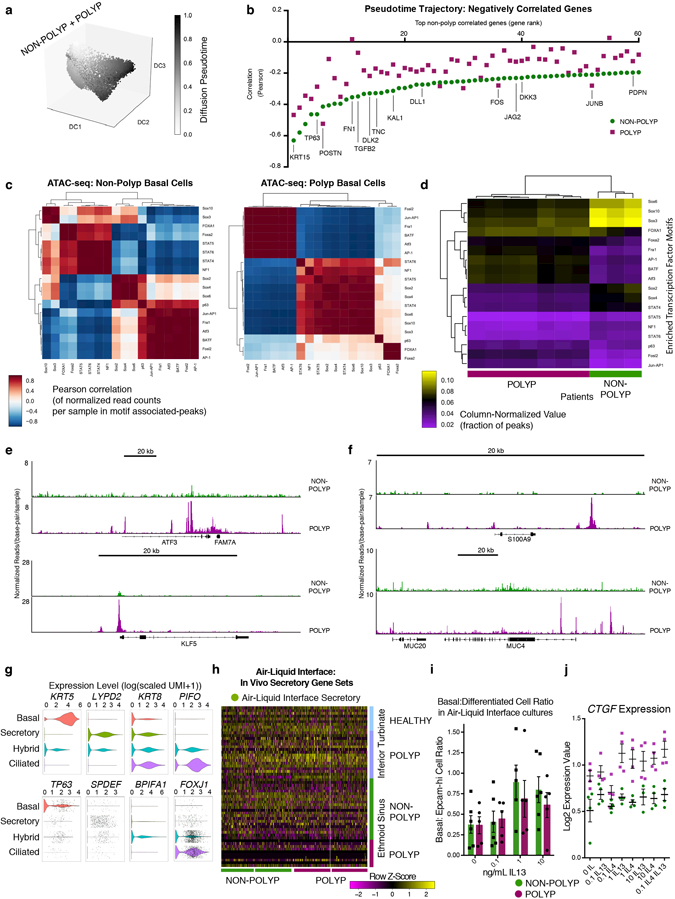
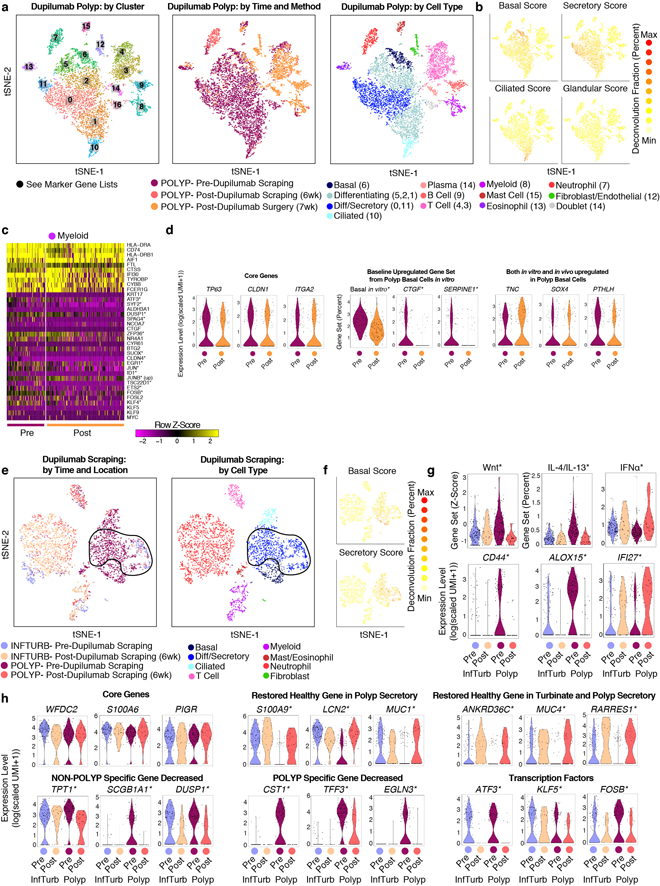
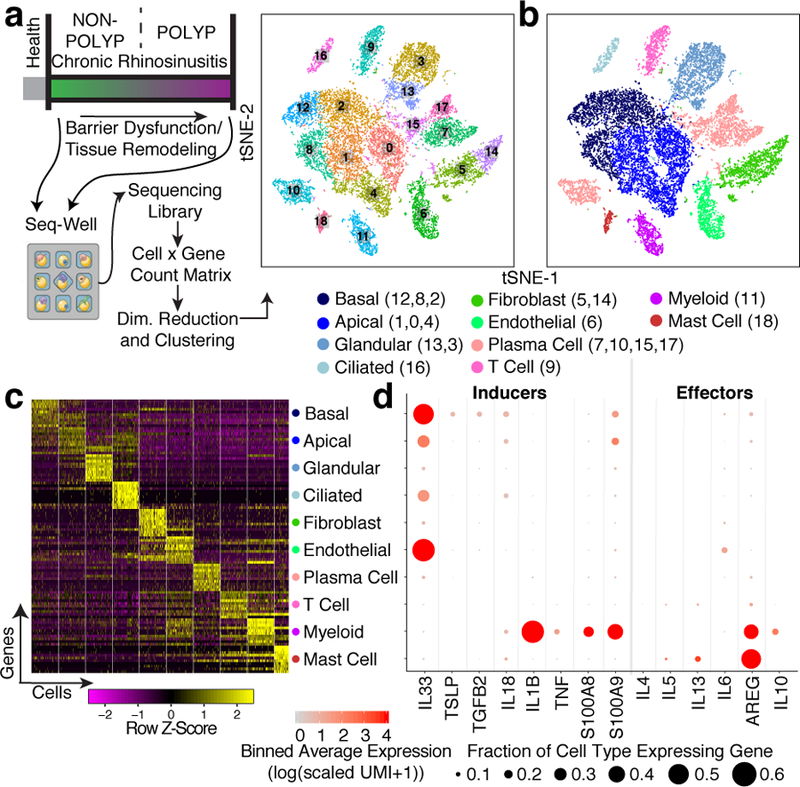
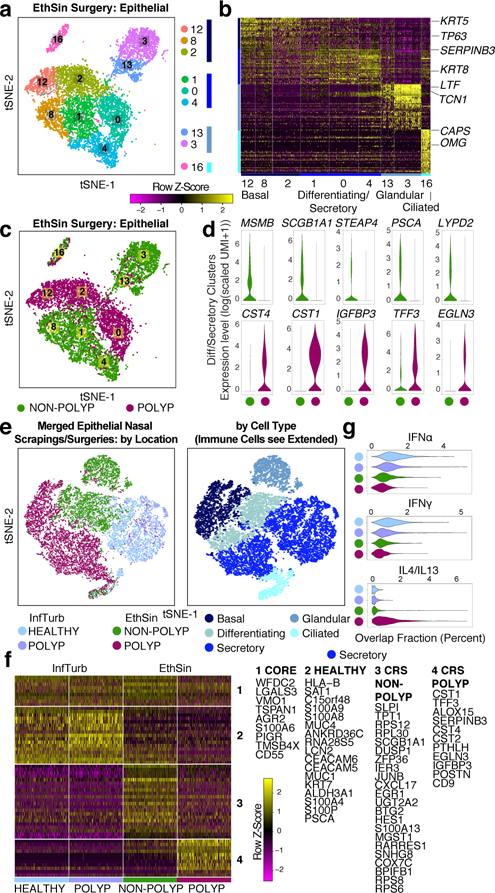
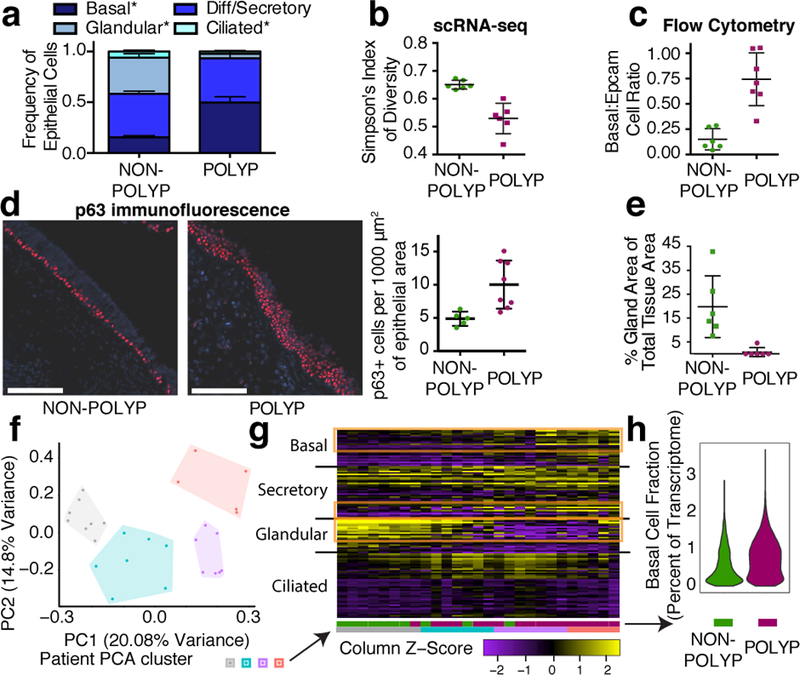
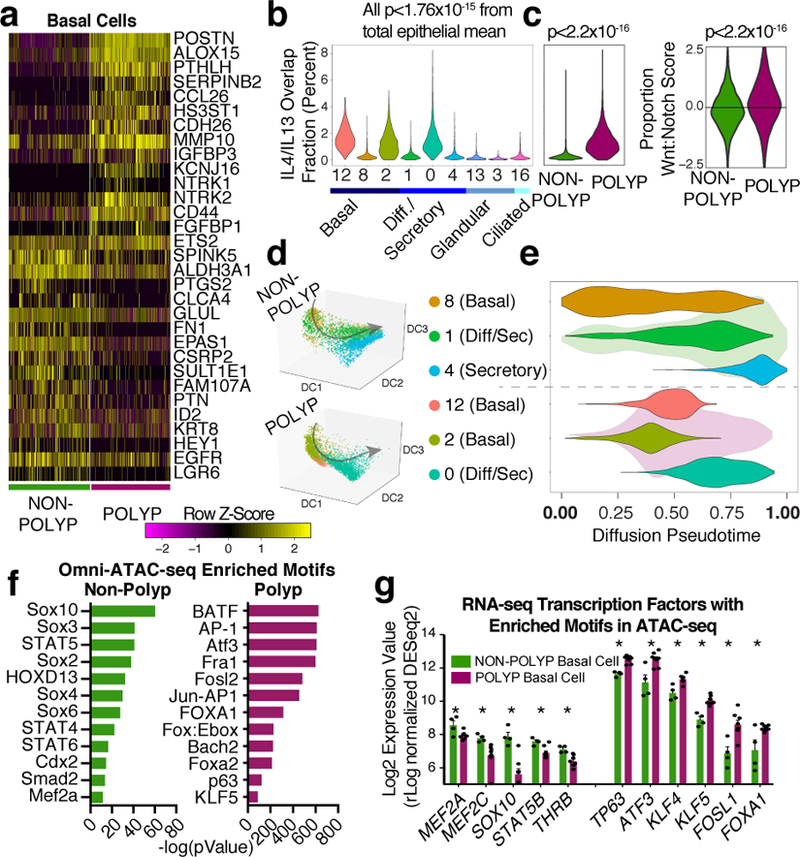
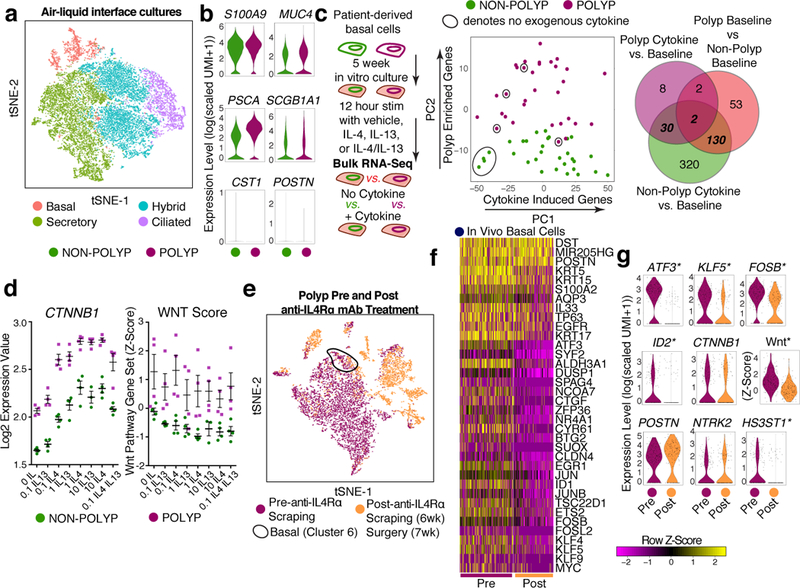
Barrier tissue dysfunction is a fundamental feature of chronic human inflammatory diseases1. Specialized subsets of epithelial cells-including secretory and ciliated cells-differentiate from basal stem cells to collectively protect the upper airway2-4. Allergic inflammation can develop from persistent activation5 of type 2 immunity6 in the upper airway, resulting in chronic rhinosinusitis, which ranges in severity from rhinitis to severe nasal polyps7. Basal cell hyperplasia is a hallmark of severe disease7-9, but it is not known how these progenitor cells2,10,11 contribute to clinical presentation and barrier tissue dysfunction in humans. Here we profile primary human surgical chronic rhinosinusitis samples (18,036 cells, n = 12) that span the disease spectrum using Seq-Well for massively parallel single-cell RNA sequencing12, report transcriptomes for human respiratory epithelial, immune and stromal cell types and subsets from a type 2 inflammatory disease, and map key mediators. By comparison with nasal scrapings (18,704 cells, n = 9), we define signatures of core, healthy, inflamed and polyp secretory cells. We reveal marked differences between the epithelial compartments of the non-polyp and polyp cellular ecosystems, identifying and validating a global reduction in cellular diversity of polyps characterized by basal cell hyperplasia, concomitant decreases in glandular cells, and phenotypic shifts in secretory cell antimicrobial expression. We detect an aberrant basal progenitor differentiation trajectory in polyps, and propose cell-intrinsic13, epigenetic14,15 and extrinsic factors11,16,17 that lock polyp basal cells into this uncommitted state. Finally, we functionally demonstrate that ex vivo cultured basal cells retain intrinsic memory of IL-4/IL-13 exposure, and test the potential for clinical blockade of the IL-4 receptor α-subunit to modify basal and secretory cell states in vivo. Overall, we find that reduced epithelial diversity stemming from functional shifts in basal cells is a key characteristic of type 2 immune-mediated barrier tissue dysfunction. Our results demonstrate that epithelial stem cells may contribute to the persistence of human disease by serving as repositories for allergic memories.
Figures
Comment in
-
Basal Instinct: Persistence of Barrier Dysfunction.Immunity. 2018 Oct 16;49(4):590-592. doi: 10.1016/j.immuni.2018.09.022. Immunity. 2018. PMID: 30332626
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL128241/HL/NHLBI NIH HHS/United States
- R01 HL095791/HL/NHLBI NIH HHS/United States
- R01 AI134989/AI/NIAID NIH HHS/United States
- R01 HL134539/HL/NHLBI NIH HHS/United States
- P30 CA014051/CA/NCI NIH HHS/United States
- DP2 GM119419/GM/NIGMS NIH HHS/United States
- R01 AI078908/AI/NIAID NIH HHS/United States
- T32 GM087237/GM/NIGMS NIH HHS/United States
- P01 AI039671/AI/NIAID NIH HHS/United States
- R33 CA202820/CA/NCI NIH HHS/United States
- R56 AI134989/AI/NIAID NIH HHS/United States
- U24 AI118672/AI/NIAID NIH HHS/United States
- R37 AI052353/AI/NIAID NIH HHS/United States
- R56 HL126554/HL/NHLBI NIH HHS/United States
- R01 AI136041/AI/NIAID NIH HHS/United States
- RM1 HG006193/HG/NHGRI NIH HHS/United States
- R01 AI138546/AI/NIAID NIH HHS/United States
- T32 AI007306/AI/NIAID NIH HHS/United States
- U19 AI095219/AI/NIAID NIH HHS/United States
- U19 AI070535/AI/NIAID NIH HHS/United States
- R01 GM081871/GM/NIGMS NIH HHS/United States
- R01 HL120952/HL/NHLBI NIH HHS/United States
- R01 HL136209/HL/NHLBI NIH HHS/United States
- U19 AI089992/AI/NIAID NIH HHS/United States
- P30 AI060354/AI/NIAID NIH HHS/United States
- K23 AI118804/AI/NIAID NIH HHS/United States
- U54 CA217377/CA/NCI NIH HHS/United States
- 1DP2OD020839/NH/NIH HHS/United States
- R01 DA046277/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
