

Depression following a traumatic brain injury: uncovering cytokine dysregulation as a pathogenic mechanism
- PMID: 30136679
- PMCID: PMC6128046
- DOI: 10.4103/1673-5374.238604
Depression following a traumatic brain injury: uncovering cytokine dysregulation as a pathogenic mechanism
Abstract
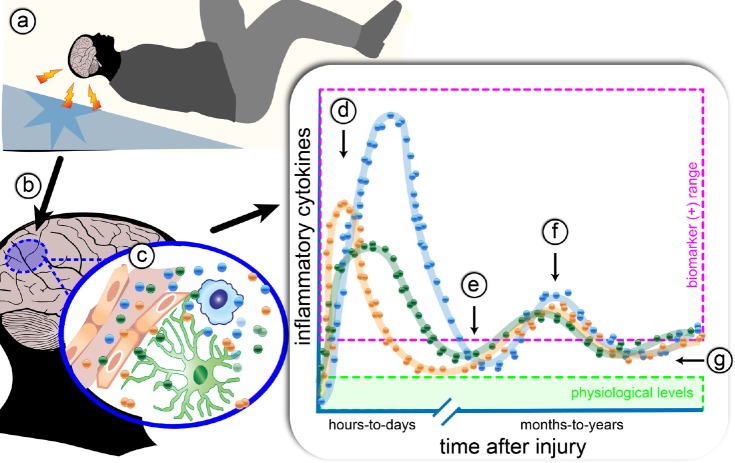
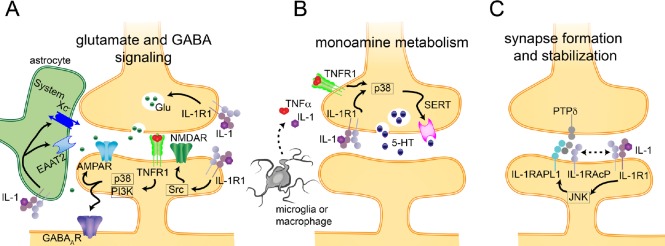
A substantial number of individuals have long-lasting adverse effects from a traumatic brain injury (TBI). Depression is one of these long-term complications that influences many aspects of life. Depression can limit the ability to return to work, and even worsen cognitive function and contribute to dementia. The mechanistic cause for the increased depression risk associated with a TBI remains to be defined. As TBI results in chronic neuroinflammation, and priming of glia to a secondary challenge, the inflammatory theory of depression provides a promising framework for investigating the cause of depression following a TBI. Increases in cytokines similar to those seen in depression in the general population are also increased following a TBI. Biomarker levels of cytokines peak within hours-to-days after the injury, yet pro-inflammatory cytokines may still be elevated above physiological levels months-to-years following TBI, which is the time frame in which post-TBI depression can persist. As tumor necrosis factor α and interleukin 1 can signal directly at the neuronal synapse, pathophysiological levels of these cytokines can detrimentally alter neuronal synaptic physiology. The purpose of this review is to outline the current evidence for the inflammatory hypothesis of depression specifically as it relates to depression following a TBI. Moreover, we will illustrate the potential synaptic mechanisms by which tumor necrosis factor α and interleukin 1 could contribute to depression. The association of inflammation with the development of depression is compelling; however, in the context of post-TBI depression, the role of inflammation is understudied. This review attempts to highlight the need to understand and treat the psychological complications of a TBI, potentially by neuroimmune modulation, as the neuropsychiatric disabilities can have a great impact on the rehabilitation from the injury, and overall quality of life.
Keywords: N-methyl-D-aspartic acid; astrocytes; chronic traumatic encephalopathy; concussion; inflammation; interleukin 1; major-depressive disorder; microglia; synaptic physiology; tumor necrosis factor α.
Conflict of interest statement
None
Figures
References
-
- Bachstetter AD, Norris CM, Sompol P, Wilcock DM, Goulding D, Neltner JH, St Clair D, Watterson DM, Van Eldik LJ. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer's disease-related pathology. J Neurosci. 2012;32:10201–10210. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
