

Comparative Genomics Approaches Accurately Predict Deleterious Variants in Plants
- PMID: 30139765
- PMCID: PMC6169392
- DOI: 10.1534/g3.118.200563
Comparative Genomics Approaches Accurately Predict Deleterious Variants in Plants
Abstract
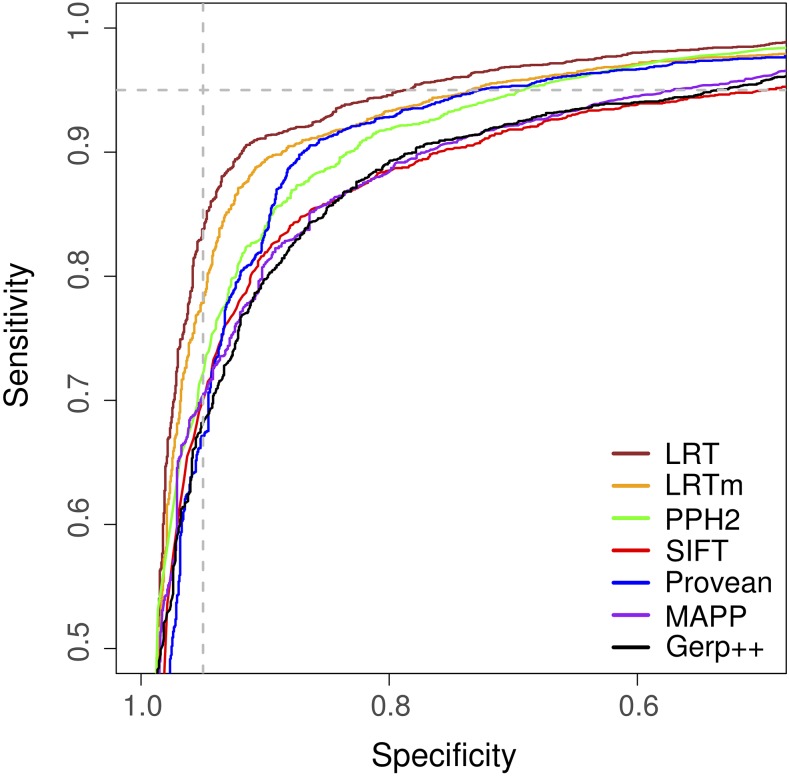
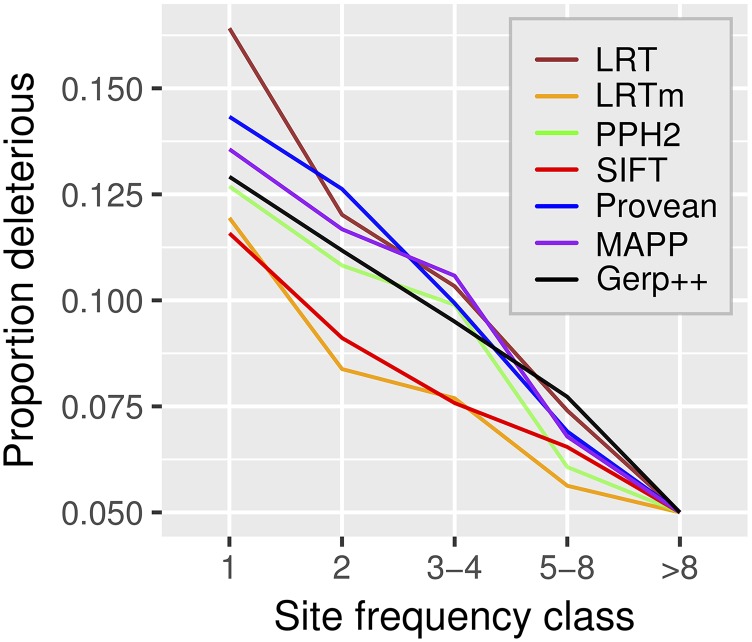
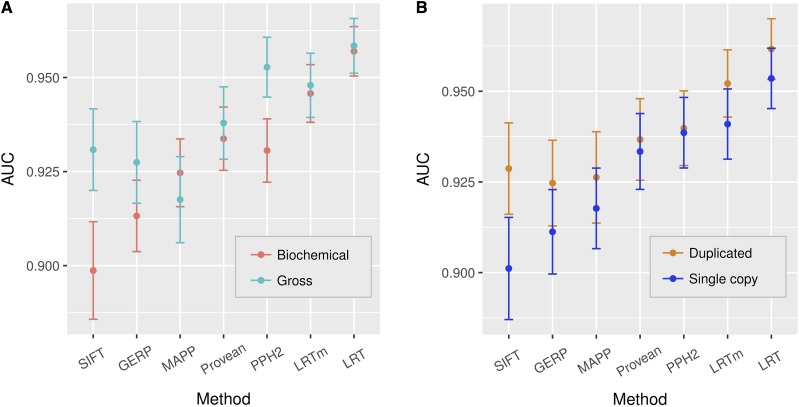
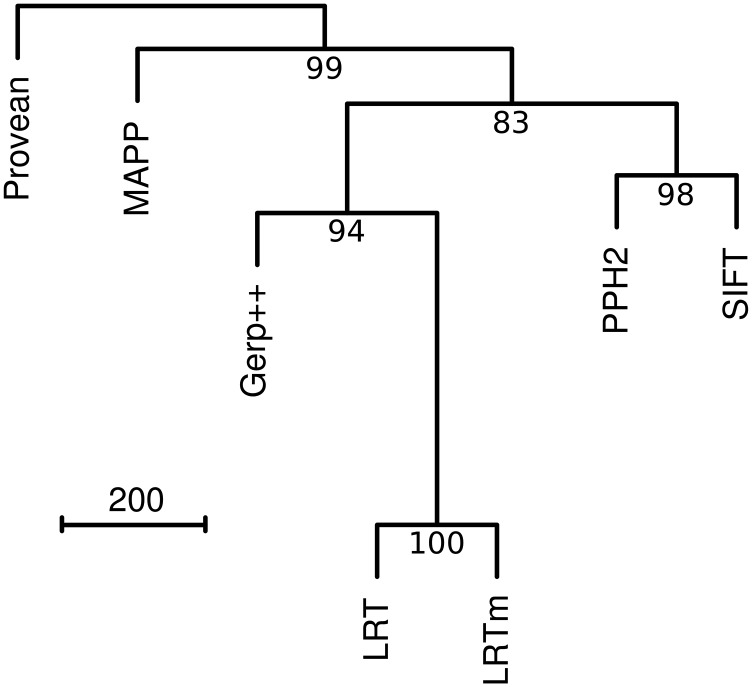
Recent advances in genome resequencing have led to increased interest in prediction of the functional consequences of genetic variants. Variants at phylogenetically conserved sites are of particular interest, because they are more likely than variants at phylogenetically variable sites to have deleterious effects on fitness and contribute to phenotypic variation. Numerous comparative genomic approaches have been developed to predict deleterious variants, but the approaches are nearly always assessed based on their ability to identify known disease-causing mutations in humans. Determining the accuracy of deleterious variant predictions in nonhuman species is important to understanding evolution, domestication, and potentially to improving crop quality and yield. To examine our ability to predict deleterious variants in plants we generated a curated database of 2,910 Arabidopsis thaliana mutants with known phenotypes. We evaluated seven approaches and found that while all performed well, their relative ranking differed from prior benchmarks in humans. We conclude that deleterious mutations can be reliably predicted in A. thaliana and likely other plant species, but that the relative performance of various approaches does not necessarily translate from one species to another.
Keywords: deleterious mutations; genome; phenotypes; training set.
Copyright © 2018 Kono et al.
Figures
References
-
- Adzhubei I., Jordan D. M., Sunyaev S. R., 2013. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. Editor. Board Jonathan Haines Al 0 7: Unit7.20 10.1002/0471142905.hg0720s76 - DOI
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources