

Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci
- PMID: 30139913
- PMCID: PMC6198826
- DOI: 10.1126/science.aar3146
Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci
Abstract
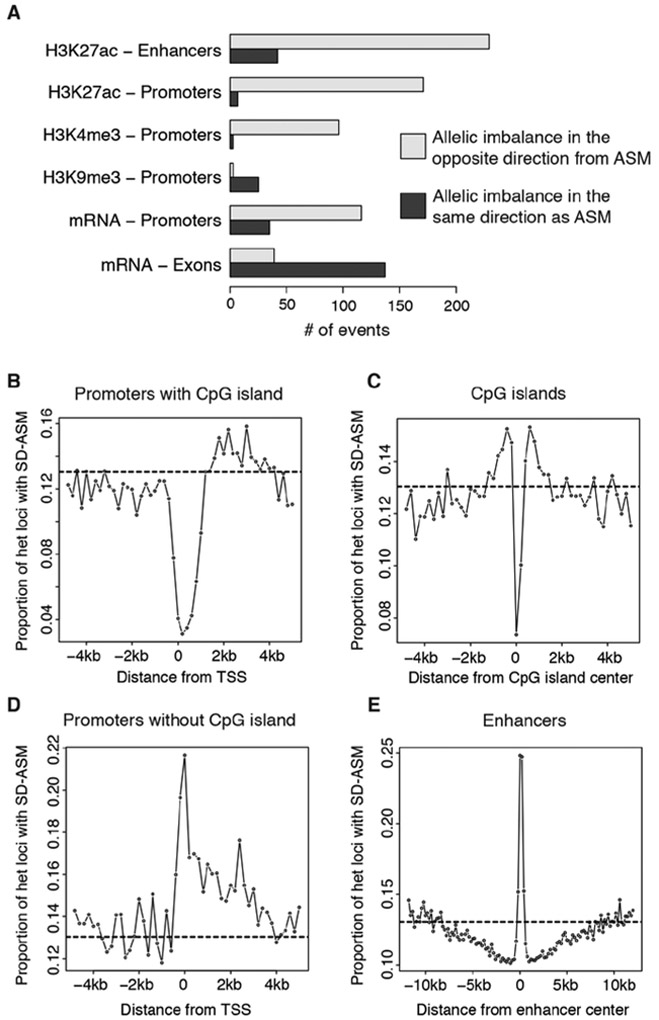
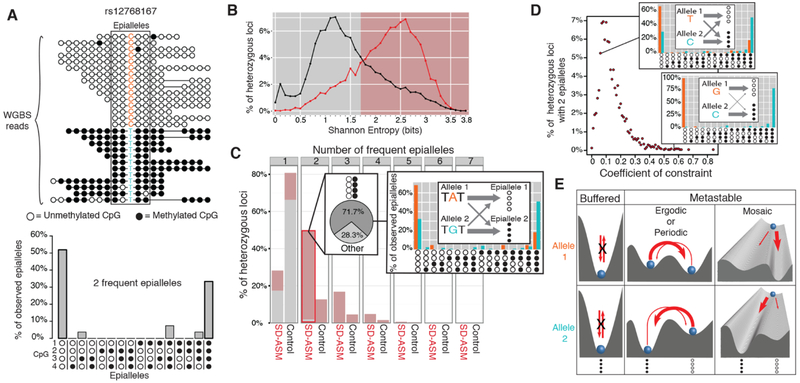
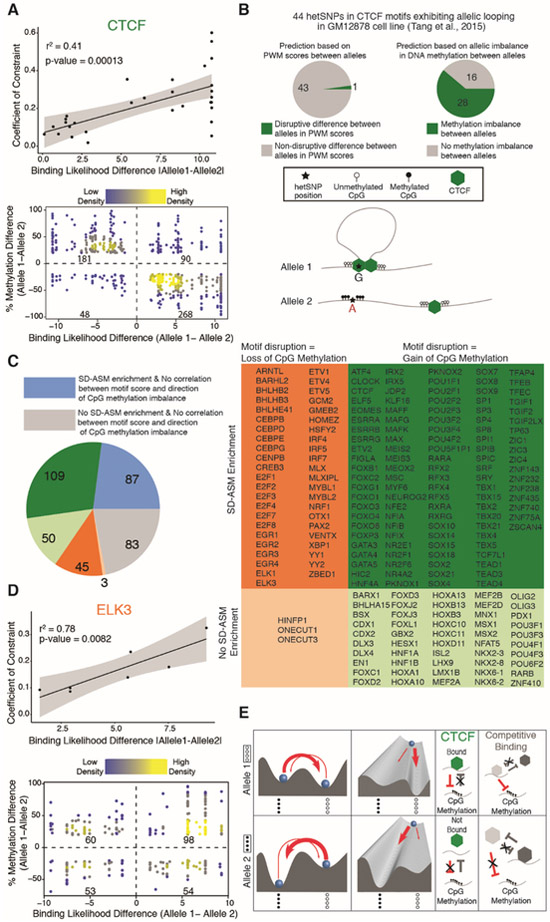
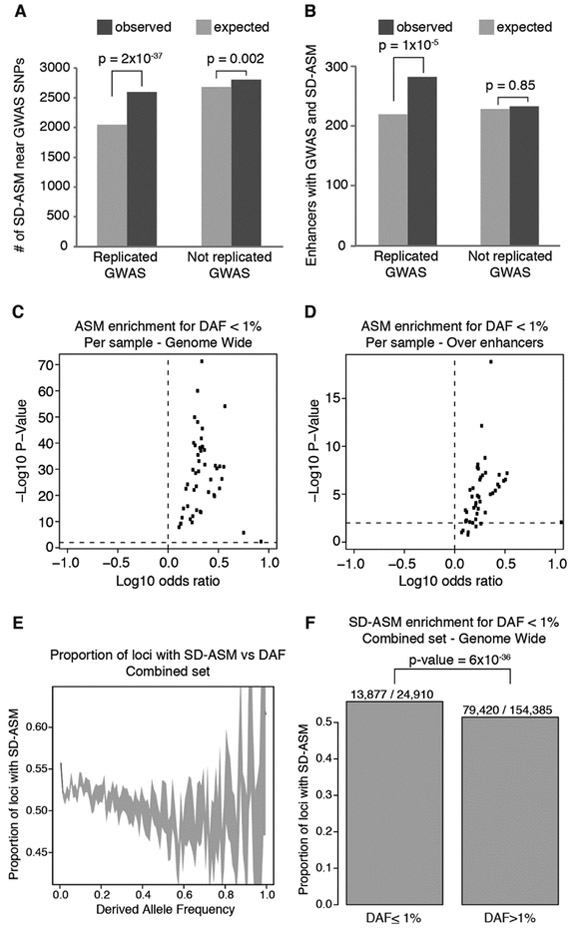
To assess the impact of genetic variation in regulatory loci on human health, we constructed a high-resolution map of allelic imbalances in DNA methylation, histone marks, and gene transcription in 71 epigenomes from 36 distinct cell and tissue types from 13 donors. Deep whole-genome bisulfite sequencing of 49 methylomes revealed sequence-dependent CpG methylation imbalances at thousands of heterozygous regulatory loci. Such loci are enriched for stochastic switching, which is defined as random transitions between fully methylated and unmethylated states of DNA. The methylation imbalances at thousands of loci are explainable by different relative frequencies of the methylated and unmethylated states for the two alleles. Further analyses provided a unifying model that links sequence-dependent allelic imbalances of the epigenome, stochastic switching at gene regulatory loci, and disease-associated genetic variation.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
References
-
- Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, Morris M, Haghighi F, Tycko B, Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet 40, 904–908 (2008). doi:10.1038/ng.174 Medline - DOI - PubMed
-
- Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, Kucera KS, Willard HF, Myers RM, Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLOS Genet. 7, e1002228 (2011). doi:10.1371/journal.pgen.1002228 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
