

Deep-coverage whole genome sequences and blood lipids among 16,324 individuals
- PMID: 30140000
- PMCID: PMC6107638
- DOI: 10.1038/s41467-018-05747-8
Deep-coverage whole genome sequences and blood lipids among 16,324 individuals
Abstract
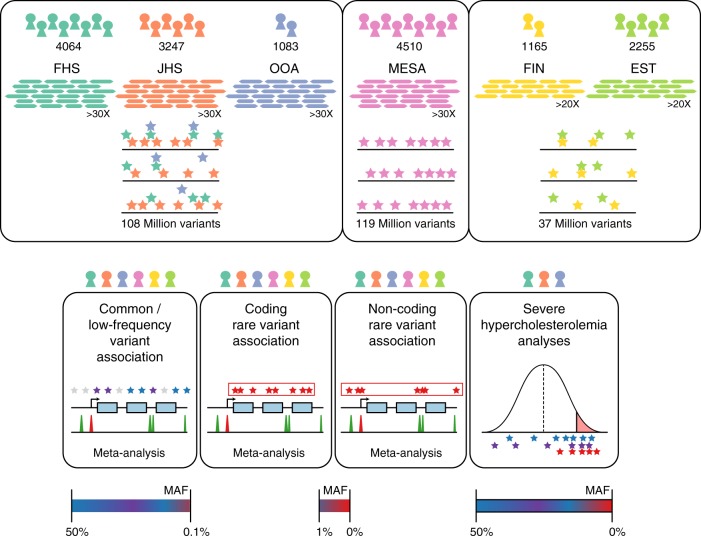
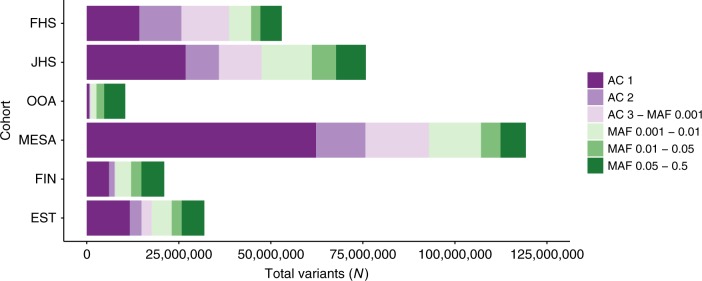
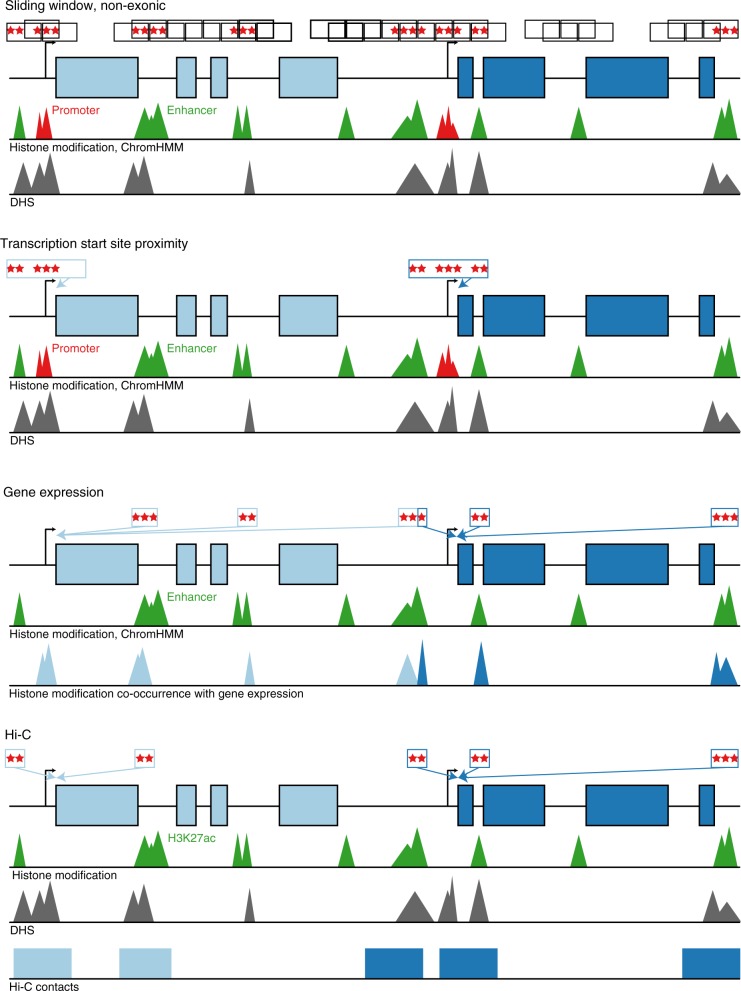
Large-scale deep-coverage whole-genome sequencing (WGS) is now feasible and offers potential advantages for locus discovery. We perform WGS in 16,324 participants from four ancestries at mean depth >29X and analyze genotypes with four quantitative traits-plasma total cholesterol, low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol, and triglycerides. Common variant association yields known loci except for few variants previously poorly imputed. Rare coding variant association yields known Mendelian dyslipidemia genes but rare non-coding variant association detects no signals. A high 2M-SNP LDL-C polygenic score (top 5th percentile) confers similar effect size to a monogenic mutation (~30 mg/dl higher for each); however, among those with severe hypercholesterolemia, 23% have a high polygenic score and only 2% carry a monogenic mutation. At these sample sizes and for these phenotypes, the incremental value of WGS for discovery is limited but WGS permits simultaneous assessment of monogenic and polygenic models to severe hypercholesterolemia.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL120393/HL/NHLBI NIH HHS/United States
- K01 HL136700/HL/NHLBI NIH HHS/United States
- R01 DK075787/DK/NIDDK NIH HHS/United States
- F30 HL107066/HL/NHLBI NIH HHS/United States
- K08 HL140203/HL/NHLBI NIH HHS/United States
- 17SDG33680041/American Heart Association (American Heart Association, Inc.)/International
- K01 HL130609/HL/NHLBI NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- U54 GM115428/GM/NIGMS NIH HHS/United States
- R01 HG002898/HG/NHGRI NIH HHS/United States
- HHSN268201100037C/HL/NHLBI NIH HHS/United States
- KL2 TR001100/TR/NCATS NIH HHS/United States
- R01 HL112064/HL/NHLBI NIH HHS/United States
- R03 HL141439/HL/NHLBI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- R01 HL109946/HL/NHLBI NIH HHS/United States
- K01 AG059898/AG/NIA NIH HHS/United States
- R01 HL121007/HL/NHLBI NIH HHS/United States
- R01 HL138737/HL/NHLBI NIH HHS/United States
- R01 HL127564/HL/NHLBI NIH HHS/United States
- K01 HL125751/HL/NHLBI NIH HHS/United States
- R01 HL142711/HL/NHLBI NIH HHS/United States
- T32 GM007205/GM/NIGMS NIH HHS/United States
- T32 HL139439/HL/NHLBI NIH HHS/United States
- U01 HL137162/HL/NHLBI NIH HHS/United States
- HHSN268201500014C/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
