

Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight
- PMID: 30140051
- PMCID: PMC6425488
- DOI: 10.1038/s41577-018-0051-1
Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight
Abstract
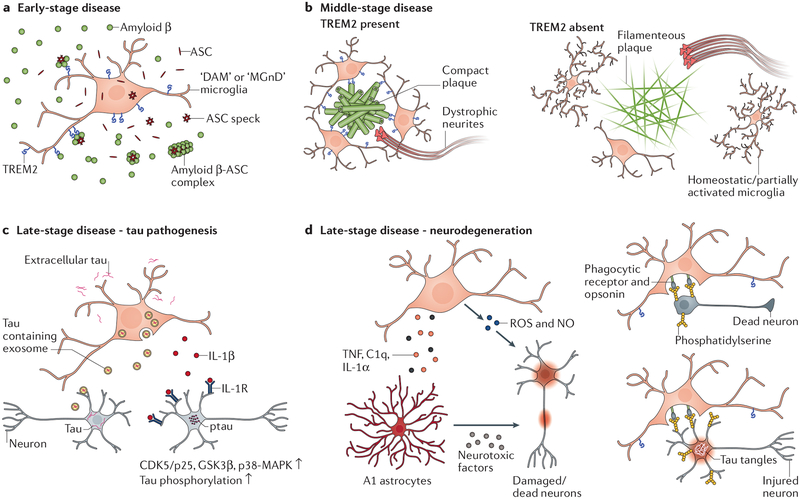
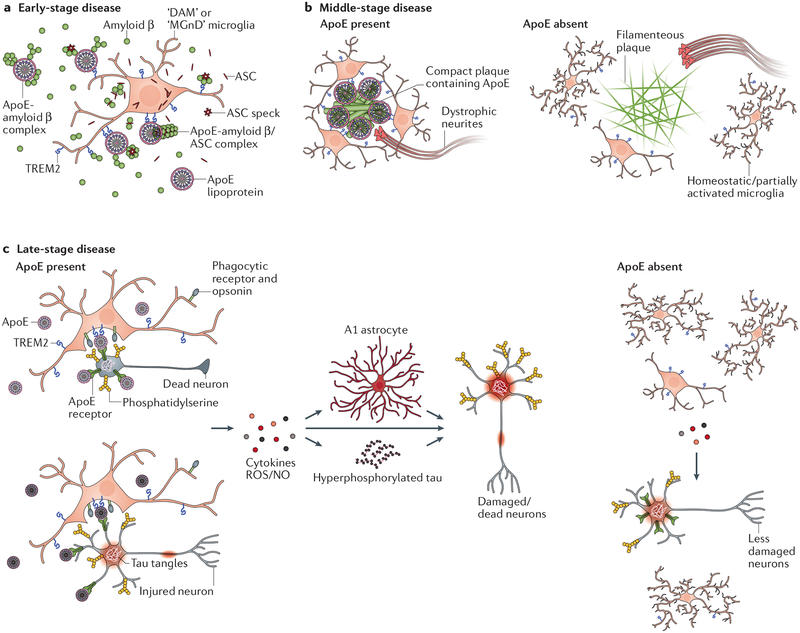
Alzheimer disease is more than a pure proteopathy. Chronic neuroinflammation stands out during the pathogenesis of the disease and in turn modulates disease progression. The central nervous system (CNS) is separated from the blood circulation by the blood-brain barrier. In Alzheimer disease, neuroinflammation heavily relies on innate immune responses that are primarily mediated by CNS-resident microglia. APOE (which encodes apolipoprotein E) is the strongest genetic risk factor for Alzheimer disease, and APOE was recently shown to affect the disease in part through its immunomodulatory function. This function of APOE is likely linked to triggering receptor expressed on myeloid cells 2 (TREM2), which is expressed by microglia in the CNS. Here, we review the rapidly growing literature on the role of disease-associated microglia, TREM2 and APOE in the pathogenesis of Alzheimer disease and present an integrated view of innate immune function in Alzheimer disease.
Conflict of interest statement
Competing interests statement
D.M.H. co-founded and is on the scientific advisory board of C2N Diagnostics. D.M.H. is on the scientific advisory board of Denali, Genentech, and Proclara. D.M.H. consults for AbbVie and Eli Lilly.
Figures
References
-
- Alzheimer A Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr 64, 146–148 (1907).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
