

CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies
- PMID: 30140266
- PMCID: PMC6094980
- DOI: 10.3389/fimmu.2018.01740
CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies
Abstract
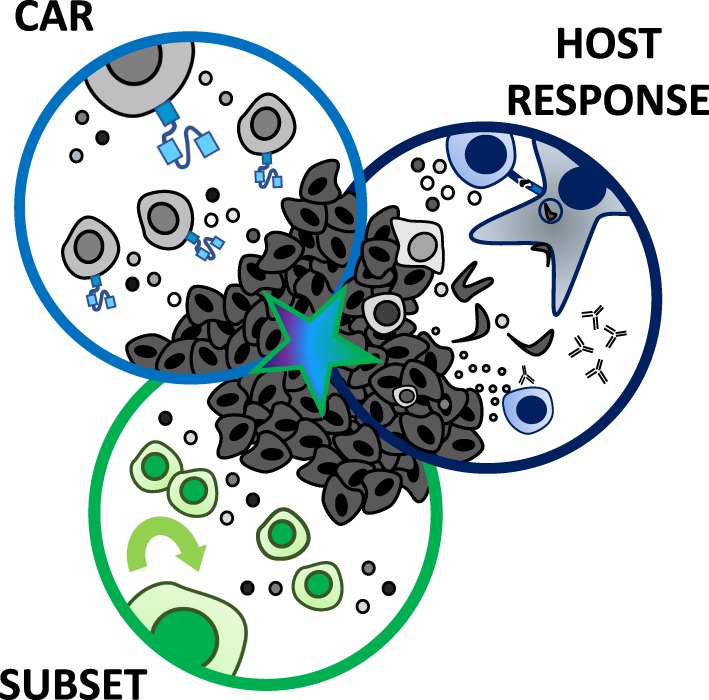
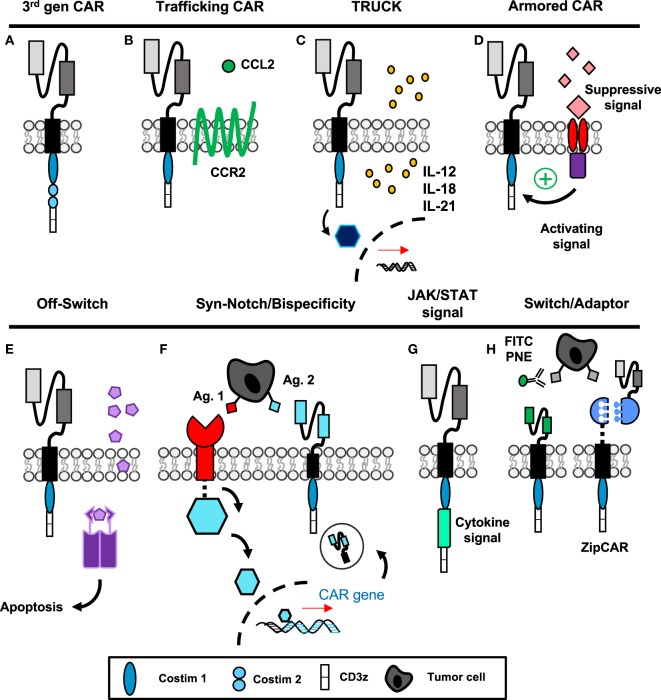
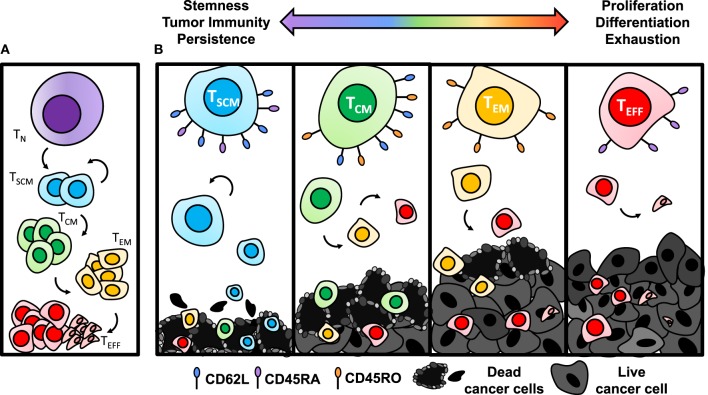
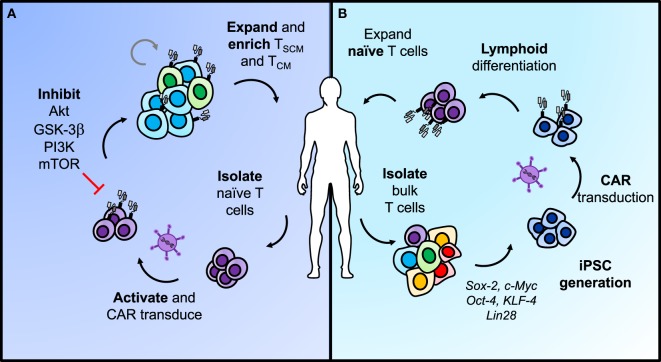
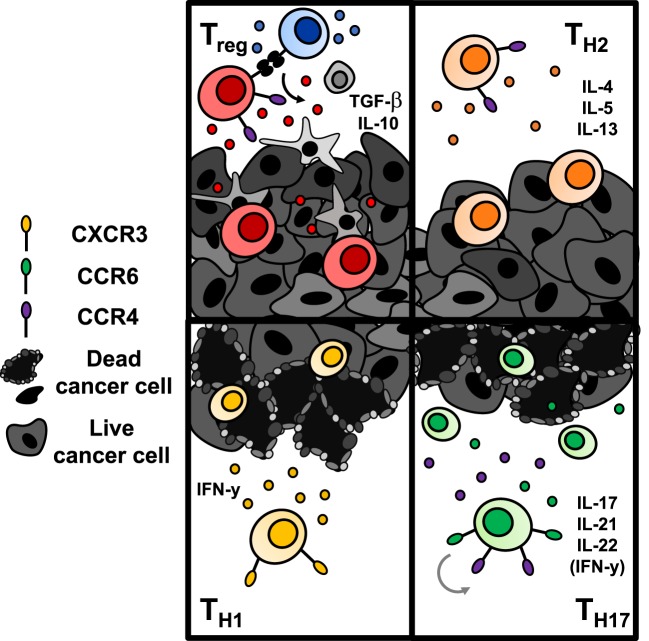
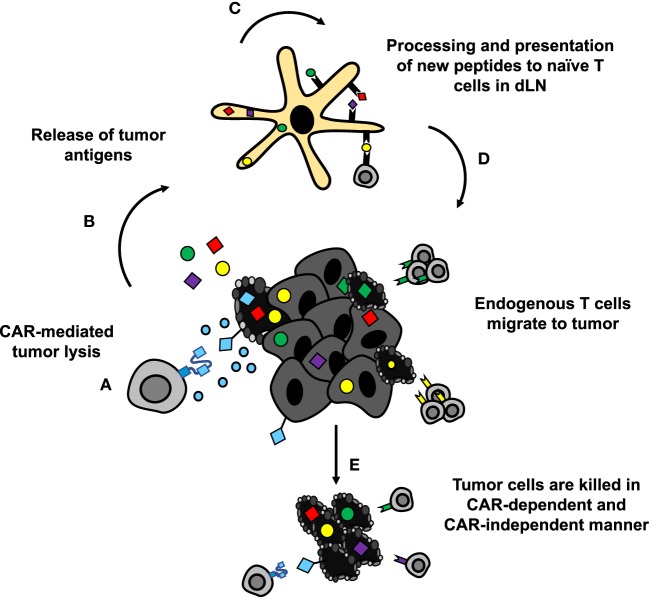
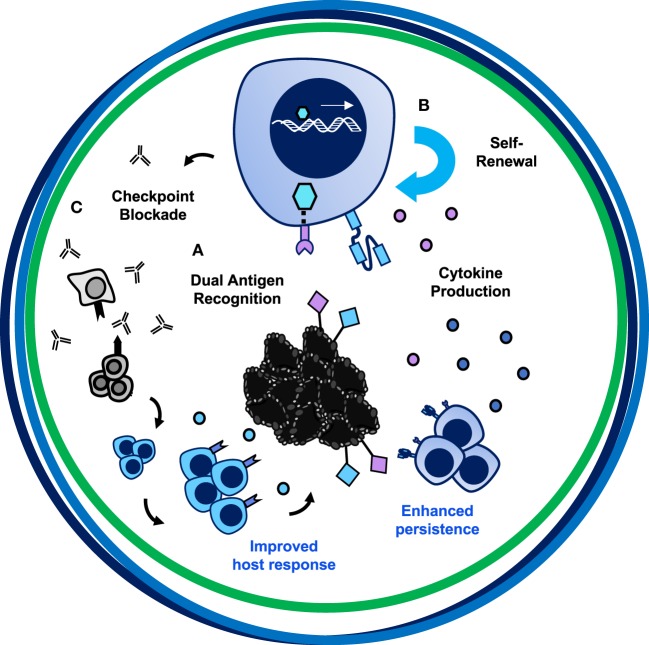
Genetic redirection of T lymphocytes with chimeric antigen receptors (CARs) has soared from treating cancers preclinically to FDA approval for hematologic malignancies and commercial-grade production scale in under 30 years. To date, solid tumors are less susceptible to CAR therapies and instead have been treated more successfully with immune checkpoint blockade or tumor-infiltrating lymphocyte therapy. Here, we discuss the current challenges in treating solid tumors with CAR T cells, and the obstacles within the host and tumor microenvironment hindering their efficacy. We present a novel three-pronged approach for enhancing the efficacy of CAR T cells whereby a single infusion product can synergize the power of an optimal CAR construct, a highly potent T cell subset, and rejuvenate the endogenous immune response to conquer therapeutically-resistant solid tumors.
Keywords: T cell; adoptive cell transfer; checkpoint; chimeric antigen receptor; solid tumor.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources