

Epigenetic Effects Induced by Methamphetamine and Methamphetamine-Dependent Oxidative Stress
- PMID: 30140365
- PMCID: PMC6081569
- DOI: 10.1155/2018/4982453
Epigenetic Effects Induced by Methamphetamine and Methamphetamine-Dependent Oxidative Stress
Abstract
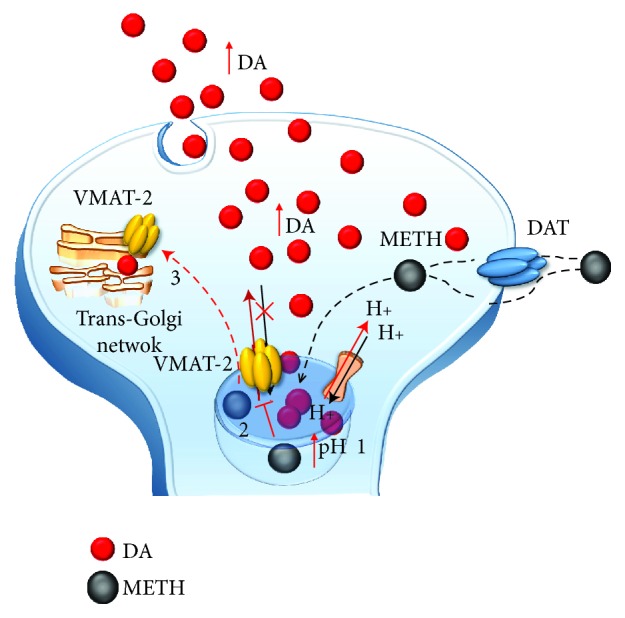
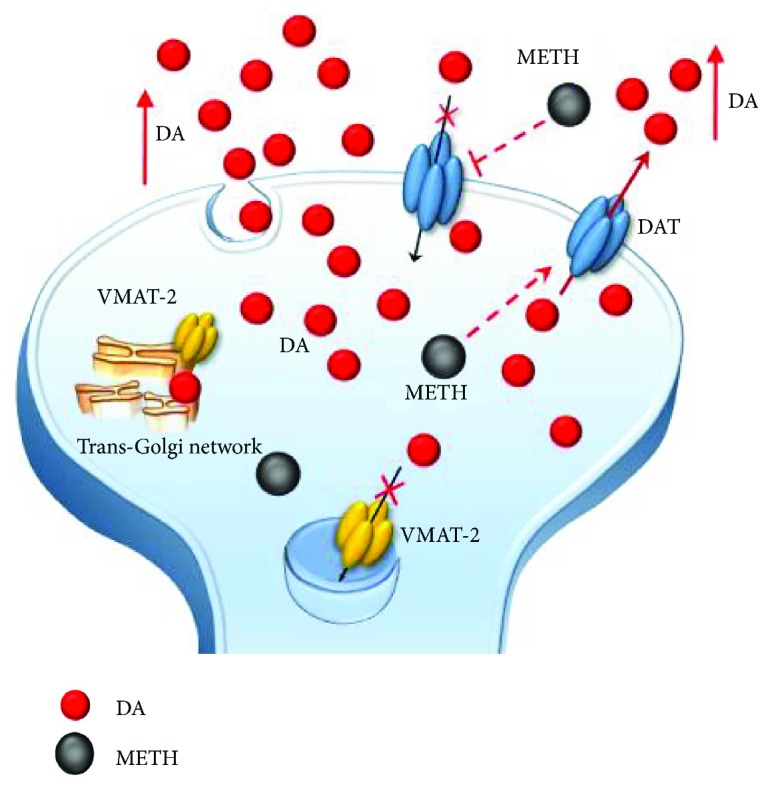
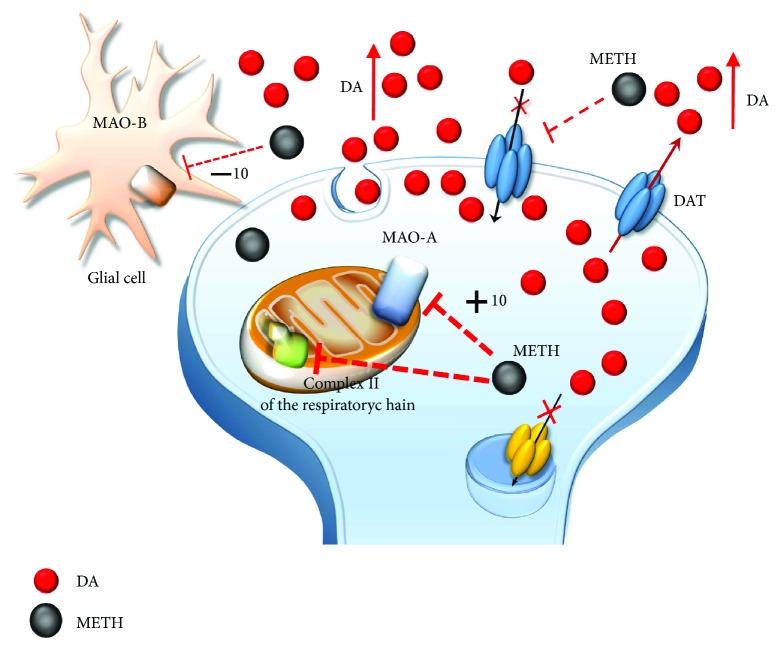
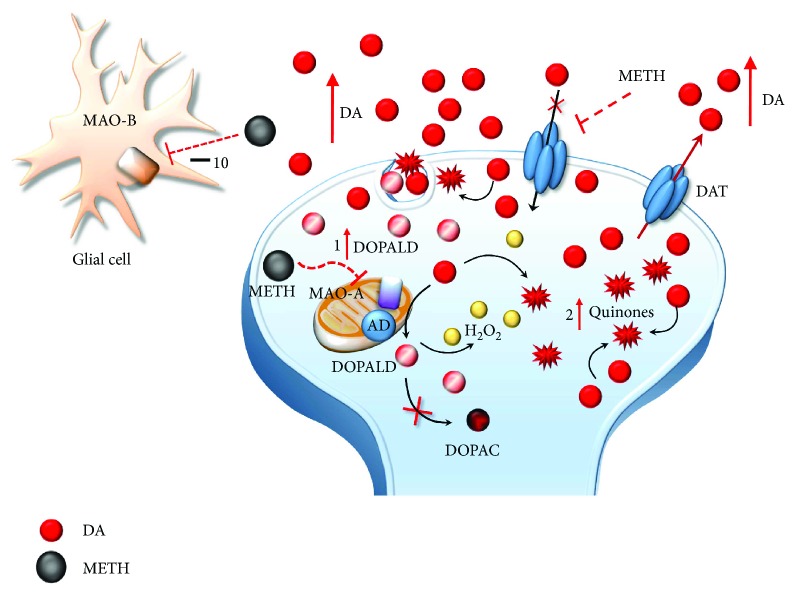
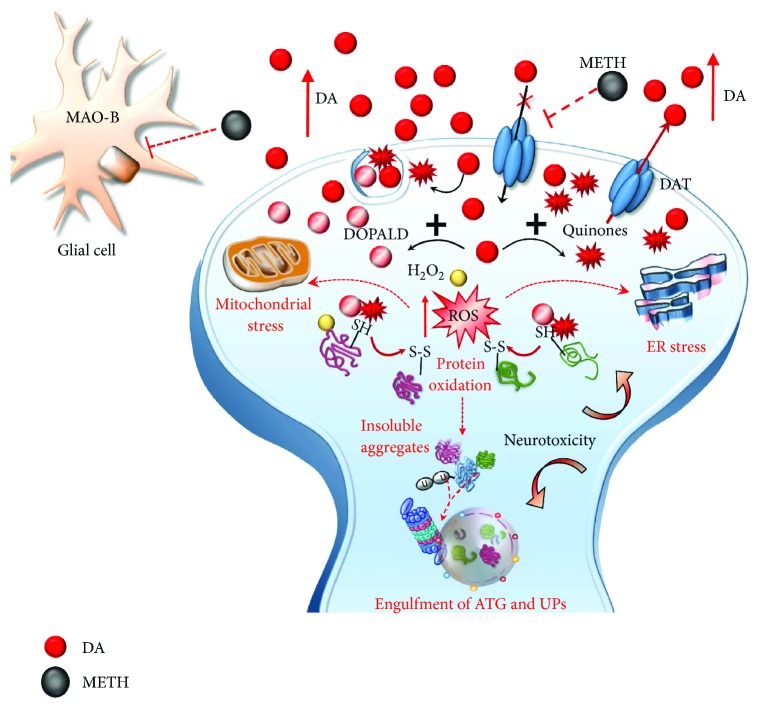
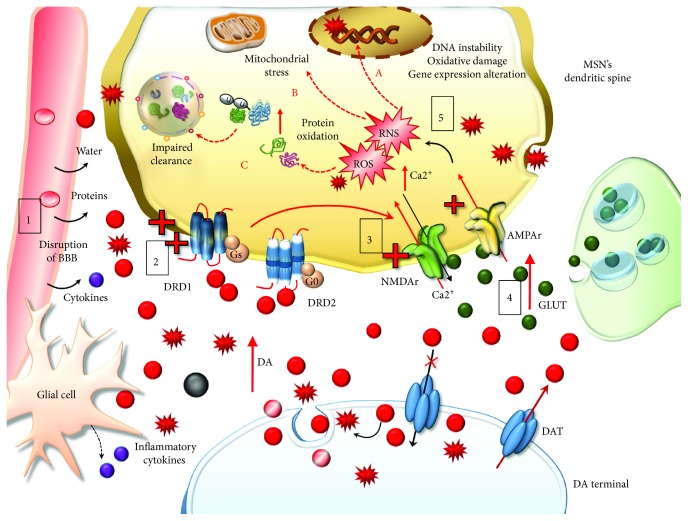
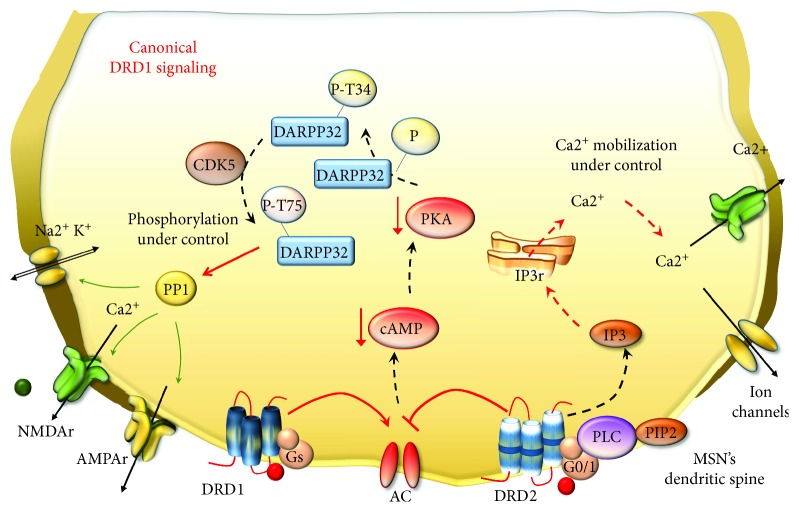
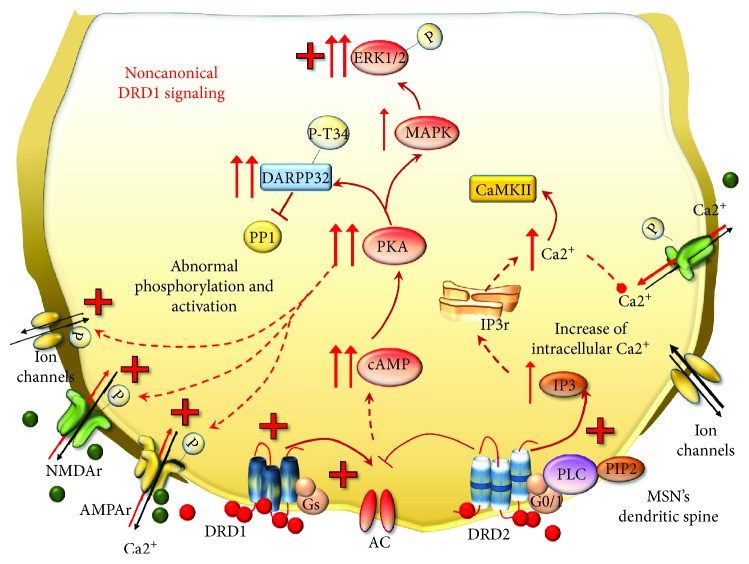
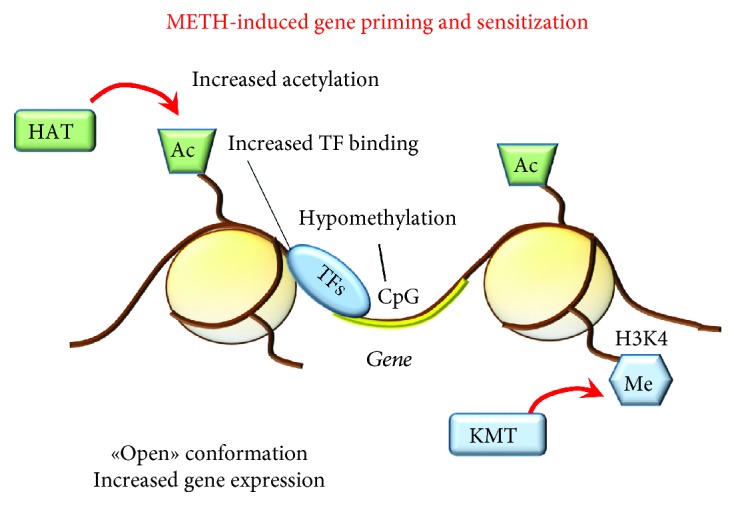
Methamphetamine is a widely abused drug, which possesses neurotoxic activity and powerful addictive effects. Understanding methamphetamine toxicity is key beyond the field of drug abuse since it allows getting an insight into the molecular mechanisms which operate in a variety of neuropsychiatric disorders. In fact, key alterations produced by methamphetamine involve dopamine neurotransmission in a way, which is reminiscent of spontaneous neurodegeneration and psychiatric schizophrenia. Thus, understanding the molecular mechanisms operated by methamphetamine represents a wide window to understand both the addicted brain and a variety of neuropsychiatric disorders. This overlapping, which is already present when looking at the molecular and cellular events promoted immediately after methamphetamine intake, becomes impressive when plastic changes induced in the brain of methamphetamine-addicted patients are considered. Thus, the present manuscript is an attempt to encompass all the molecular events starting at the presynaptic dopamine terminals to reach the nucleus of postsynaptic neurons to explain how specific neurotransmitters and signaling cascades produce persistent genetic modifications, which shift neuronal phenotype and induce behavioral alterations. A special emphasis is posed on disclosing those early and delayed molecular events, which translate an altered neurotransmitter function into epigenetic events, which are derived from the translation of postsynaptic noncanonical signaling into altered gene regulation. All epigenetic effects are considered in light of their persistent changes induced in the postsynaptic neurons including sensitization and desensitization, priming, and shift of neuronal phenotype.
Figures
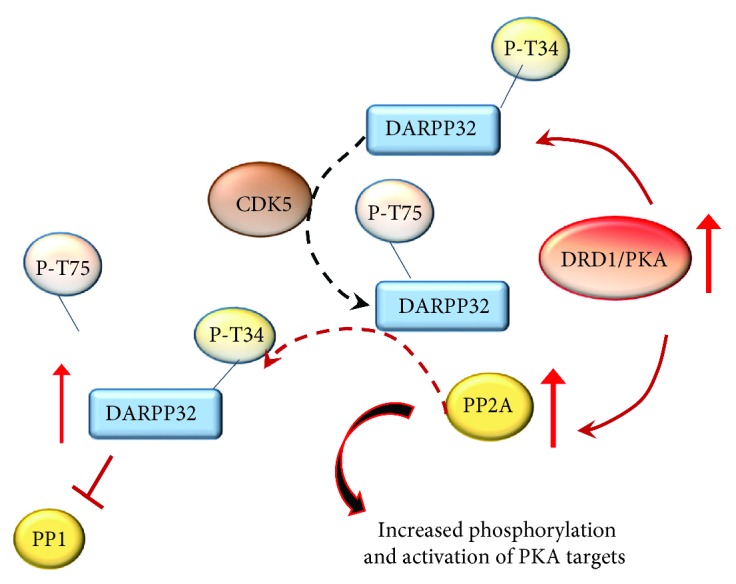
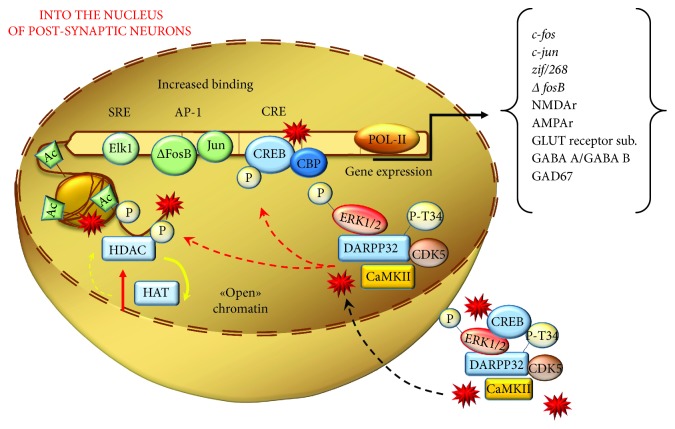
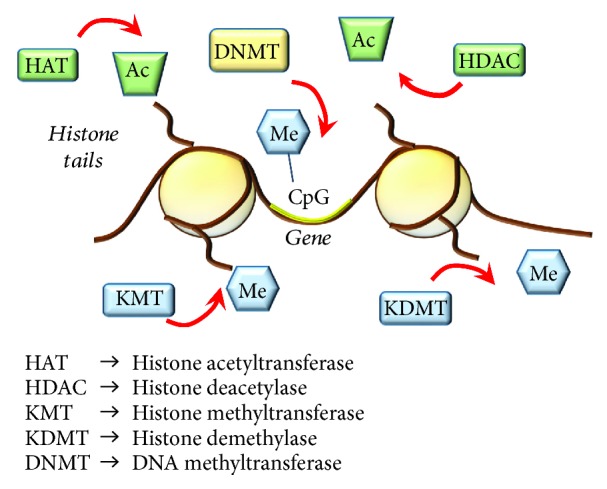
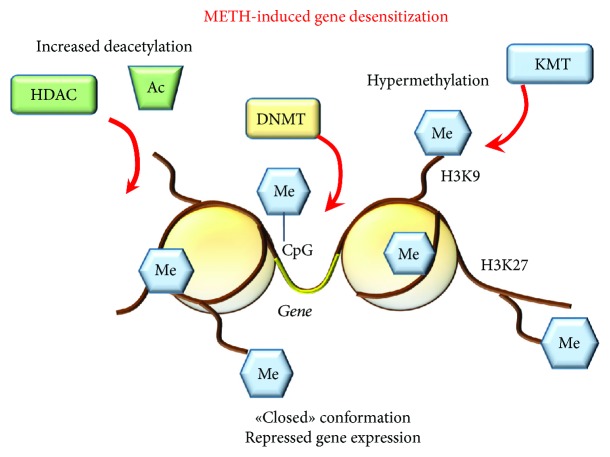
References
-
- Schepers R. J., Oyler J. M., Joseph R. E., Jr., Cone E. J., Moolchan E. T., Huestis M. A. Methamphetamine and amphetamine pharmacokinetics in oral fluid and plasma after controlled oral methamphetamine administration to human volunteers. Clinical Chemistry. 2003;49(1):121–132. doi: 10.1373/49.1.121. - DOI - PubMed
-
- Homer B. D., Solomon T. M., Moeller R. W., Mascia A., DeRaleau L., Halkitis P. N. Methamphetamine abuse and impairment of social functioning: a review of the underlying neurophysiological causes and behavioral implications. Psychological Bulletin. 2008;134(2):301–310. doi: 10.1037/0033-2909.134.2.301. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
