

Enter the Matrix: Factorization Uncovers Knowledge from Omics
- PMID: 30143323
- PMCID: PMC6309559
- DOI: 10.1016/j.tig.2018.07.003
Enter the Matrix: Factorization Uncovers Knowledge from Omics
Abstract
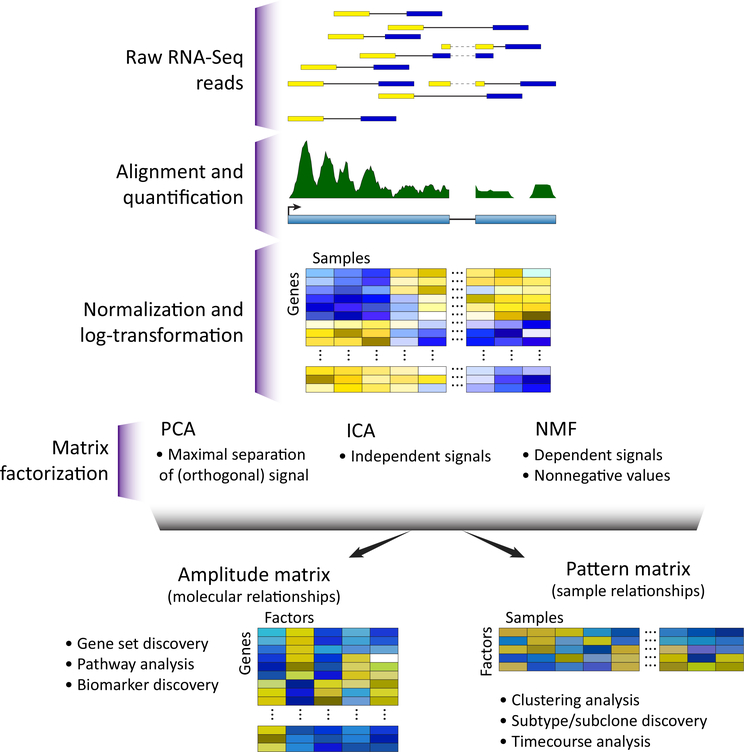
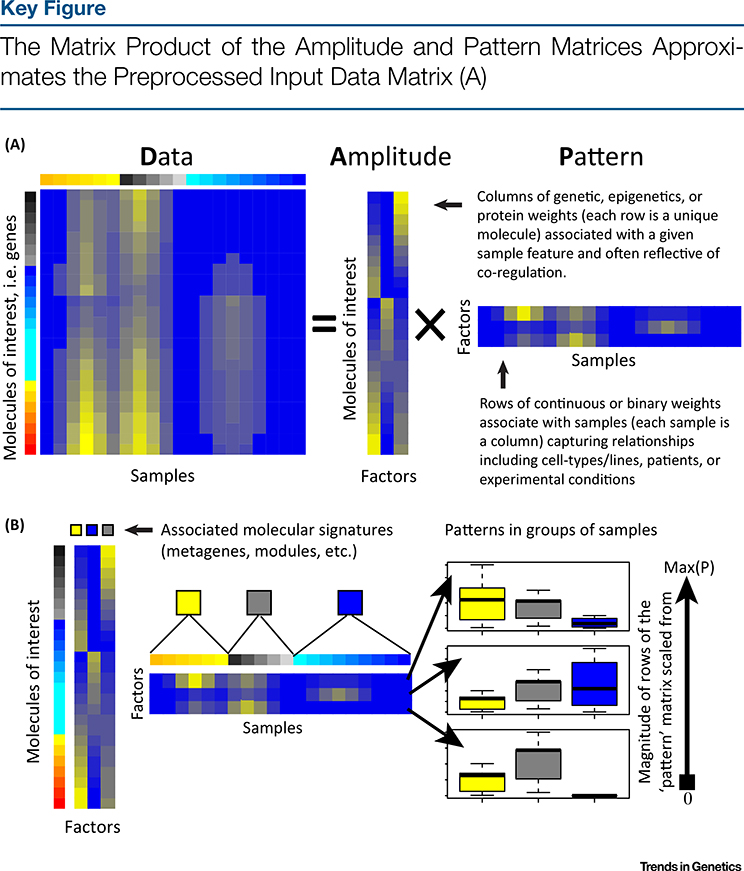
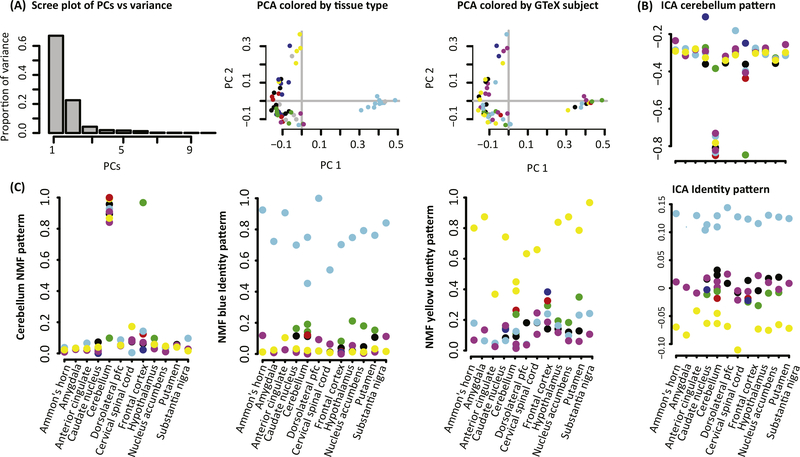
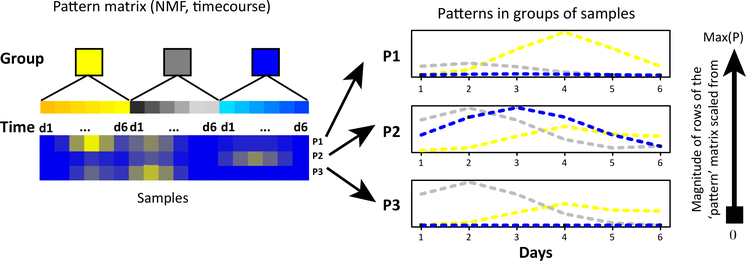
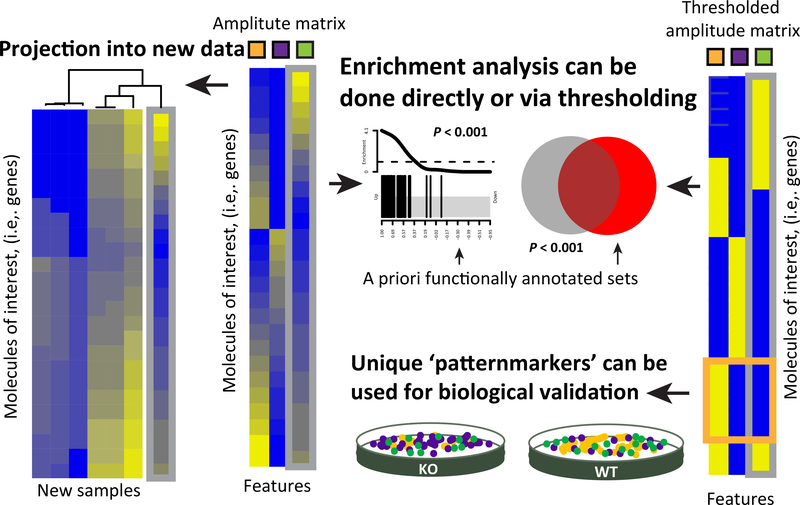
Omics data contain signals from the molecular, physical, and kinetic inter- and intracellular interactions that control biological systems. Matrix factorization (MF) techniques can reveal low-dimensional structure from high-dimensional data that reflect these interactions. These techniques can uncover new biological knowledge from diverse high-throughput omics data in applications ranging from pathway discovery to timecourse analysis. We review exemplary applications of MF for systems-level analyses. We discuss appropriate applications of these methods, their limitations, and focus on the analysis of results to facilitate optimal biological interpretation. The inference of biologically relevant features with MF enables discovery from high-throughput data beyond the limits of current biological knowledge - answering questions from high-dimensional data that we have not yet thought to ask.
Keywords: deconvolution; dimension reduction; genomics; matrix factorization; single cell; unsupervised learning.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Figures
References
-
- Bell G et al. (2009) Beyond the data deluge. Science 323, 1297–1298 - PubMed
-
- Sagoff M (2012) Data deluge and the human microbiome project. Issues Sci. Technol 28 http://issues.org/28-4/sagoff-3/
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
