

Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency
- PMID: 30143481
- PMCID: PMC6170168
- DOI: 10.1084/jem.20180628
Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency
Abstract
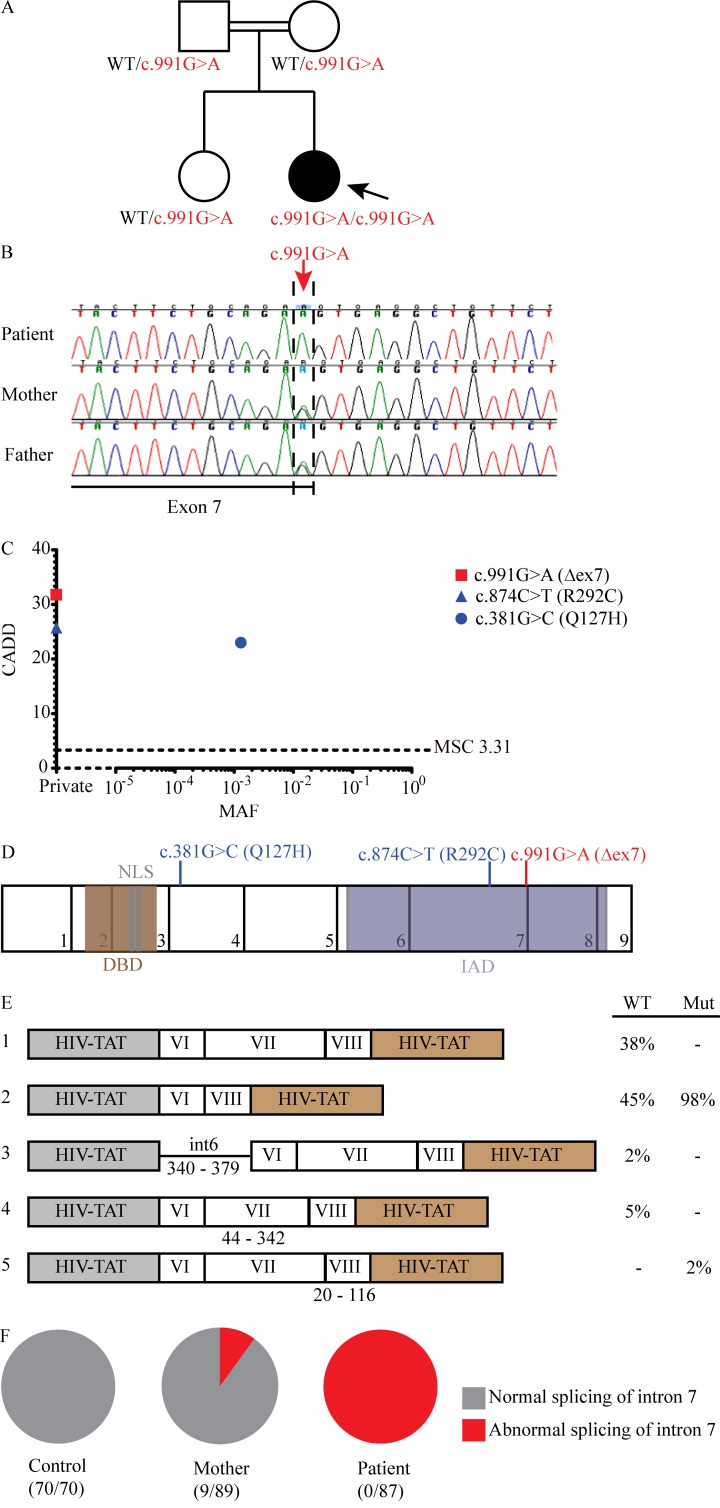
Life-threatening pulmonary influenza can be caused by inborn errors of type I and III IFN immunity. We report a 5-yr-old child with severe pulmonary influenza at 2 yr. She is homozygous for a loss-of-function IRF9 allele. Her cells activate gamma-activated factor (GAF) STAT1 homodimers but not IFN-stimulated gene factor 3 (ISGF3) trimers (STAT1/STAT2/IRF9) in response to IFN-α2b. The transcriptome induced by IFN-α2b in the patient's cells is much narrower than that of control cells; however, induction of a subset of IFN-stimulated gene transcripts remains detectable. In vitro, the patient's cells do not control three respiratory viruses, influenza A virus (IAV), parainfluenza virus (PIV), and respiratory syncytial virus (RSV). These phenotypes are rescued by wild-type IRF9, whereas silencing IRF9 expression in control cells increases viral replication. However, the child has controlled various common viruses in vivo, including respiratory viruses other than IAV. Our findings show that human IRF9- and ISGF3-dependent type I and III IFN responsive pathways are essential for controlling IAV.
© 2018 Hernandez et al.
Figures





References
-
- Andrews S.2010. FastQC: a quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
