

Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract
- PMID: 30143558
- PMCID: PMC6115658
- DOI: 10.1681/ASN.2017121265
Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract
Abstract
Background: Congenital anomalies of the kidney and urinary tract (CAKUT) are the most prevalent cause of kidney disease in the first three decades of life. Previous gene panel studies showed monogenic causation in up to 12% of patients with CAKUT.
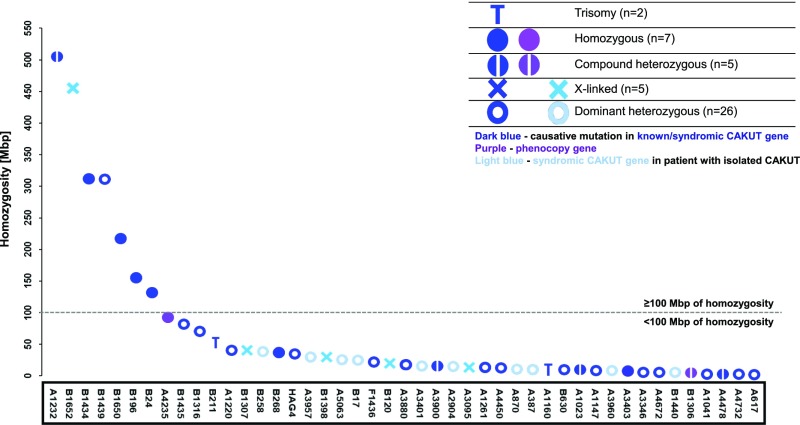
Methods: We applied whole-exome sequencing to analyze the genotypes of individuals from 232 families with CAKUT, evaluating for mutations in single genes known to cause human CAKUT and genes known to cause CAKUT in mice. In consanguineous or multiplex families, we additionally performed a search for novel monogenic causes of CAKUT.
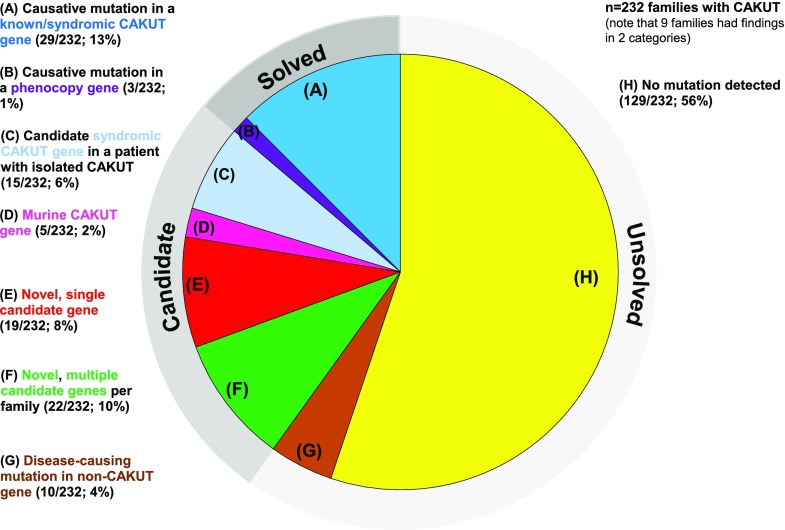
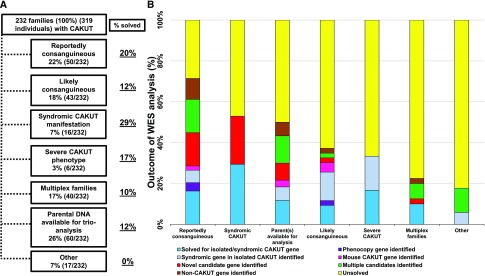
Results: In 29 families (13%), we detected a causative mutation in a known gene for isolated or syndromic CAKUT that sufficiently explained the patient's CAKUT phenotype. In three families (1%), we detected a mutation in a gene reported to cause a phenocopy of CAKUT. In 15 of 155 families with isolated CAKUT, we detected deleterious mutations in syndromic CAKUT genes. Our additional search for novel monogenic causes of CAKUT in consanguineous and multiplex families revealed a potential single, novel monogenic CAKUT gene in 19 of 232 families (8%).
Conclusions: We identified monogenic mutations in a known human CAKUT gene or CAKUT phenocopy gene as the cause of disease in 14% of the CAKUT families in this study. Whole-exome sequencing provides an etiologic diagnosis in a high fraction of patients with CAKUT and will provide a new basis for the mechanistic understanding of CAKUT.
Keywords: Congenital Anomalies of the Kidney and Urinary Tract (CAKUT); Vesico-ureteral Reflux (VUR); Whole Exome Sequencing (WES); monogenic disease causation; renal developmental gene.
Copyright © 2018 by the American Society of Nephrology.
Figures
References
-
- Chesnaye N, Bonthuis M, Schaefer F, Groothoff JW, Verrina E, Heaf JG, et al.: ESPN/ERA–EDTA registry : Demographics of paediatric renal replacement therapy in Europe: A report of the ESPN/ERA-EDTA registry. Pediatr Nephrol 29: 2403–2410, 2014 - PubMed
-
- North American Pediatric Renal Transplant Cooperative Study: NAPRTCS 2008 Annual Report, Rockville, MD, The EMMES Corporation, 2008
-
- Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, et al.: Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet 9: 358–364, 1995 - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- P20 DK116191/DK/NIDDK NIH HHS/United States
- R01 DK088767/DK/NIDDK NIH HHS/United States
- P30 DK079310/DK/NIDDK NIH HHS/United States
- R01 DK076683/DK/NIDDK NIH HHS/United States
- U54 HG006504/HG/NHGRI NIH HHS/United States
- R01 DK096238/DK/NIDDK NIH HHS/United States
- T32 DK007726/DK/NIDDK NIH HHS/United States
- S10 OD030363/OD/NIH HHS/United States
- R01 DK103184/DK/NIDDK NIH HHS/United States
- R01 DK115574/DK/NIDDK NIH HHS/United States
- R01 DK078226/DK/NIDDK NIH HHS/United States
- R56 DK096238/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
