

Molecular mechanisms of viral oncogenesis in humans
- PMID: 30143749
- PMCID: PMC6336458
- DOI: 10.1038/s41579-018-0064-6
Molecular mechanisms of viral oncogenesis in humans
Abstract
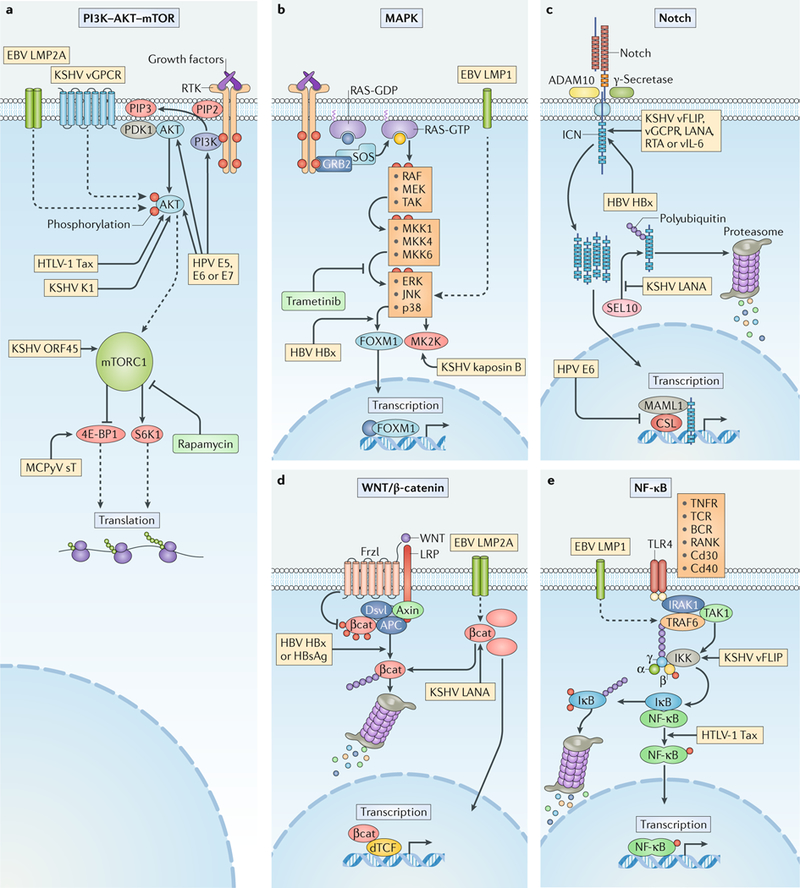
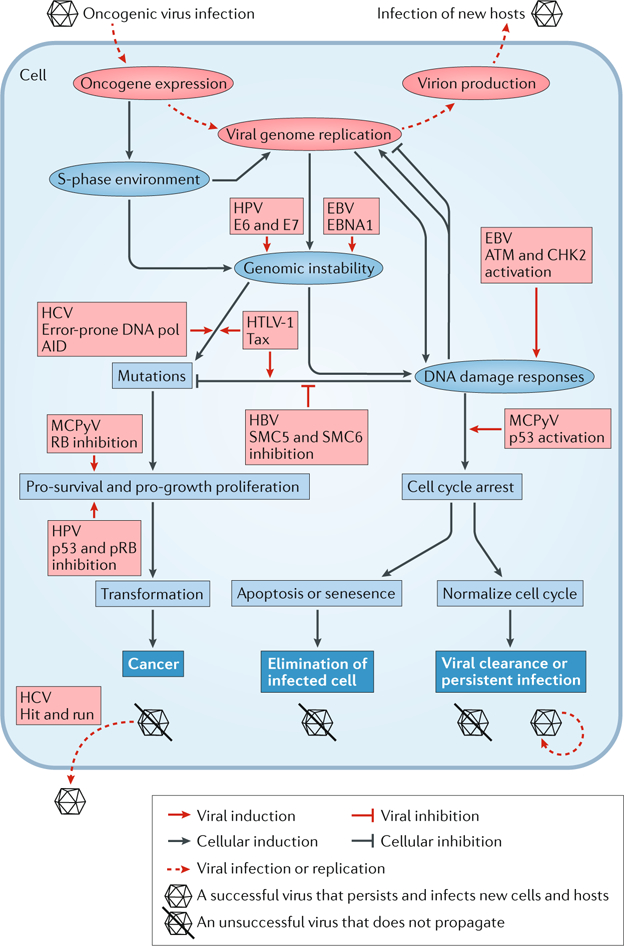
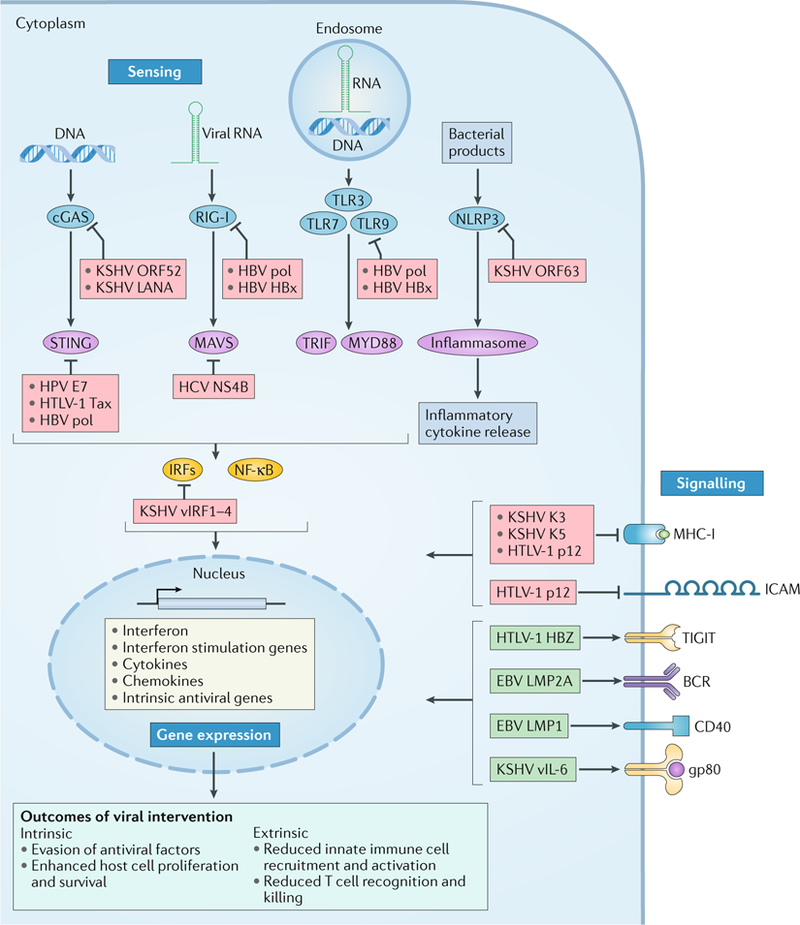
Viral infection is a major contributor to the global cancer burden. Recent advances have revealed that seven known oncogenic viruses promote tumorigenesis through shared host cell targets and pathways. A comprehensive understanding of the principles of viral oncogenesis may enable the identification of unknown infectious aetiologies of cancer and the development of therapeutic or preventive strategies for virus-associated cancers. In this Review, we discuss the molecular mechanisms of viral oncogenesis in humans. We highlight recent advances in understanding how viral manipulation of host cellular signalling, DNA damage responses, immunity and microRNA targets promotes the initiation and development of cancer.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- zur Hausen H & de Villiers EM Cancer “causation” by infections — individual contributions and synergistic networks. Semin. Oncol 41, 860–875 (2014). - PubMed
-
- Epstein MA, Achong BG & Barr YM Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1, 702–703 (1964). - PubMed
-
-
Chang Y et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869 (1994).
In this study, representational difference analysis identifies KSHV as a new human herpesvirus associated with Kaposi sarcoma.
-
-
- Doorbar J Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci 110, 525–541 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
