

Inhibiting pyrimidine biosynthesis impairs Ebola virus replication through depletion of nucleoside pools and activation of innate immune responses
- PMID: 30144461
- PMCID: PMC6436837
- DOI: 10.1016/j.antiviral.2018.08.012
Inhibiting pyrimidine biosynthesis impairs Ebola virus replication through depletion of nucleoside pools and activation of innate immune responses
Abstract
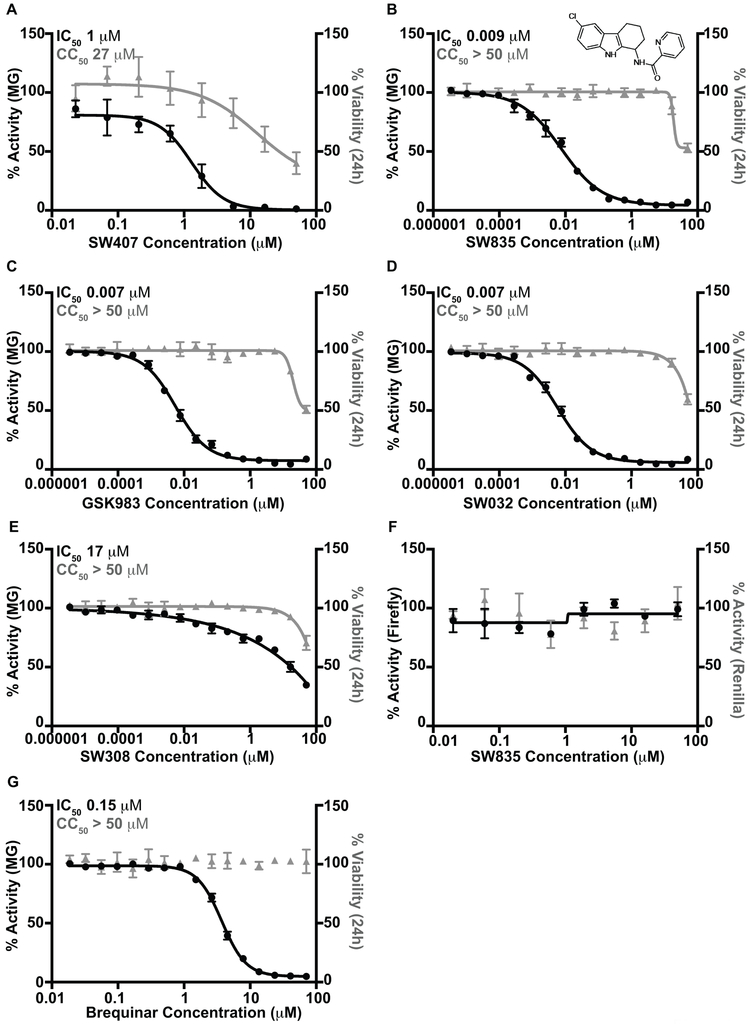
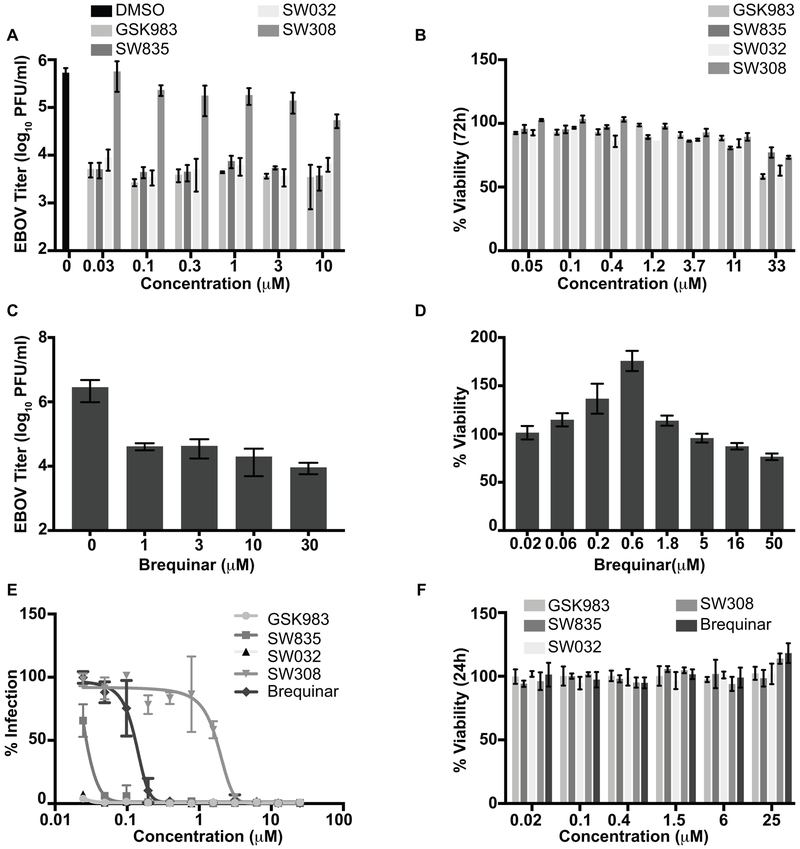
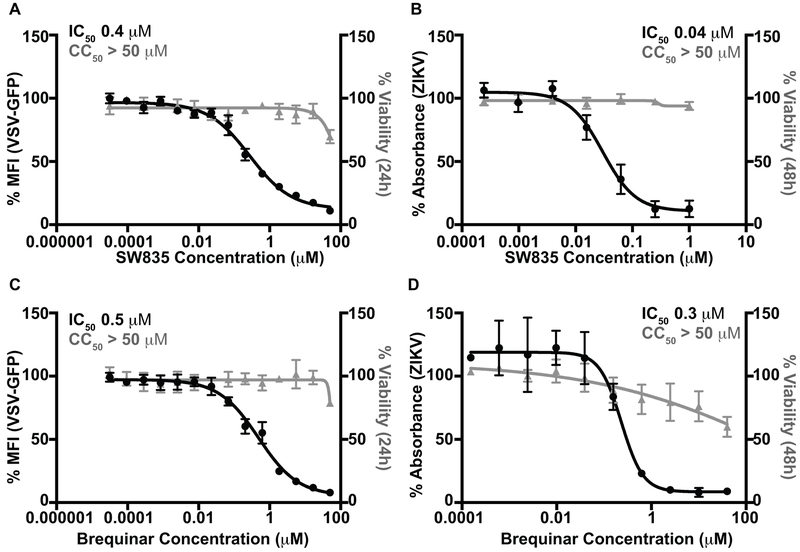
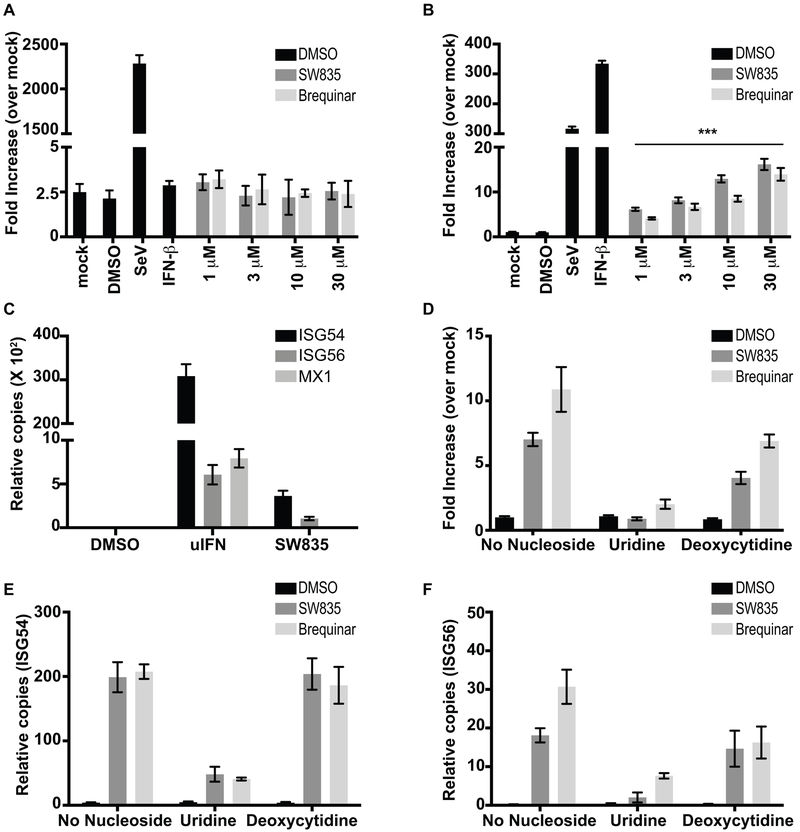
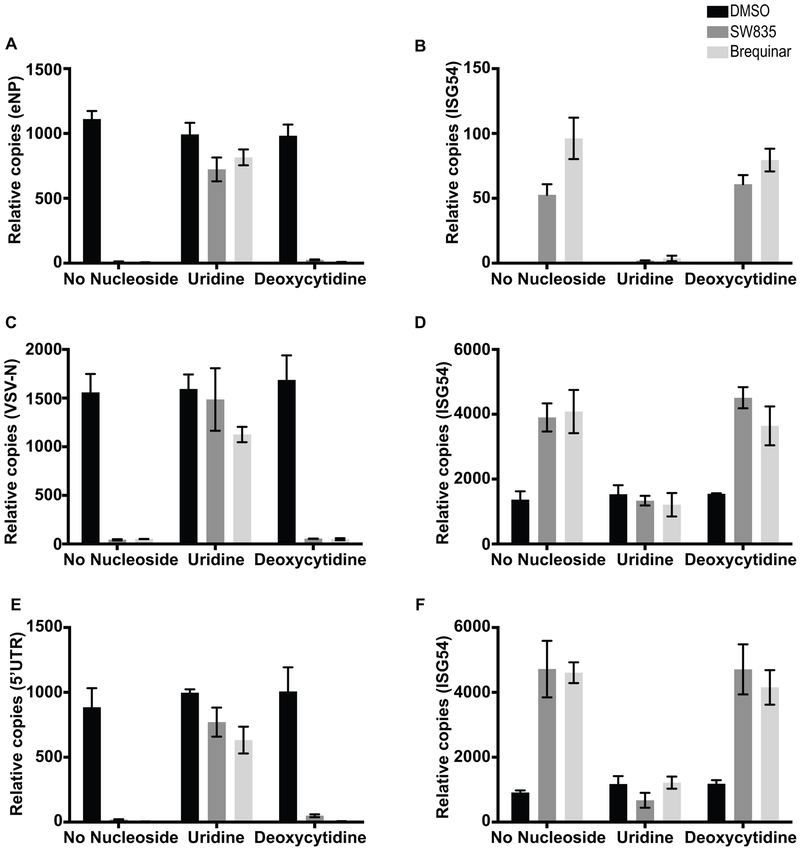
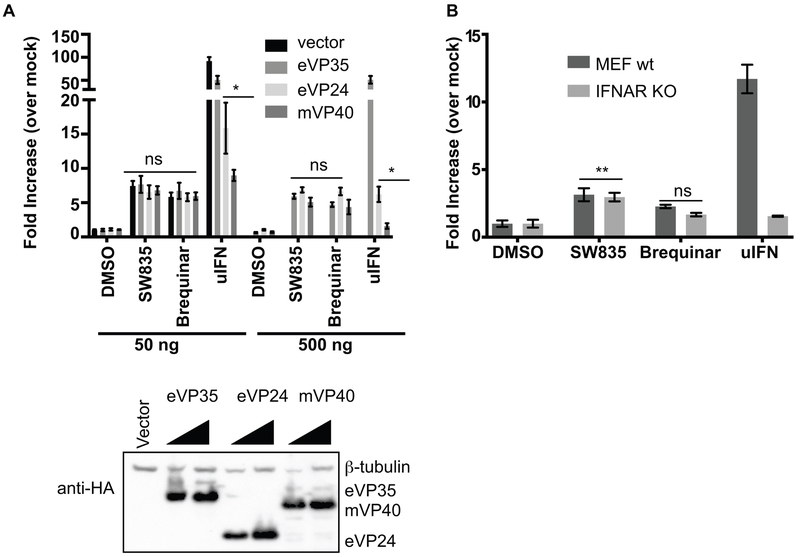
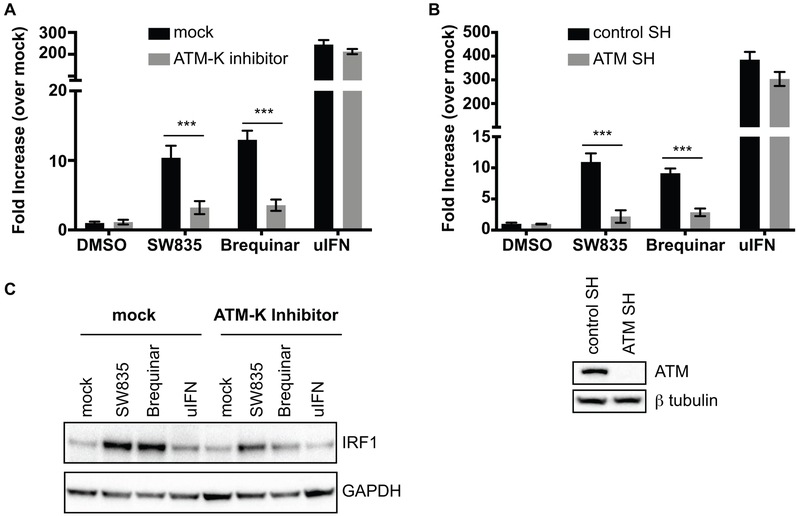
Specific host pathways that may be targeted therapeutically to inhibit the replication of Ebola virus (EBOV) and other emerging viruses remain incompletely defined. A screen of 200,000 compounds for inhibition of an EBOV minigenome (MG) assay that measures the function of the viral polymerase complex identified as hits several compounds with an amino-tetrahydrocarbazole scaffold. This scaffold was structurally similar to GSK983, a compound previously described as having broad-spectrum antiviral activity due to its impairing de novo pyrimidine biosynthesis through inhibition of dihydroorotate dehydrogenase (DHODH). We generated compound SW835, the racemic version of GSK983 and demonstrated that SW835 and brequinar, another DHODH inhibitor, potently inhibit the MG assay and the replication of EBOV, vesicular stomatitis virus (VSV) and Zika (ZIKV) in vitro. Nucleoside and deoxynucleoside supplementation studies demonstrated that depletion of pyrimidine pools contributes to antiviral activity of these compounds. As reported for other DHODH inhibitors, SW835 and brequinar also induced expression of interferon stimulated genes (ISGs). ISG induction was demonstrated to occur without production of IFNα/β and independently of the IFNα receptor and was not blocked by EBOV-encoded suppressors of IFN signaling pathways. Furthermore, we demonstrated that transcription factor IRF1 is required for this ISG induction, and that IRF1 induction requires the DNA damage response kinase ATM. Therefore, de novo pyrimidine biosynthesis is critical for the replication of EBOV and other RNA viruses and inhibition of this pathway activates an ATM and IRF1-dependent innate immune response that subverts EBOV immune evasion functions.
Copyright © 2018 Elsevier B.V. All rights reserved.
Figures
References
-
- Afonso CL, Amarasinghe GK, Banyai K, Bao Y, Basler CF, Bavari S, Bejerman N, Blasdell KR, Briand FX, Briese T, Bukreyev A, Calisher CH, Chandran K, Cheng J, Clawson AN, Collins PL, Dietzgen RG, Dolnik O, Domier LL, Durrwald R, Dye JM, Easton AJ, Ebihara H, Farkas SL, Freitas-Astua J, Formenty P, Fouchier RA, Fu Y, Ghedin E, Goodin MM, Hewson R, Horie M, Hyndman TH, Jiang D, Kitajima EW, Kobinger GP, Kondo H, Kurath G, Lamb RA, Lenardon S, Leroy EM, Li CX, Lin XD, Liu L, Longdon B, Marton S, Maisner A, Muhlberger E, Netesov SV, Nowotny N, Patterson JL, Payne SL, Paweska JT, Randall RE, Rima BK, Rota P, Rubbenstroth D, Schwemmle M, Shi M, Smither SJ, Stenglein MD, Stone DM, Takada A, Terregino C, Tesh RB, Tian JH, Tomonaga K, Tordo N, Towner JS, Vasilakis N, Verbeek M, Volchkov VE, Wahl-Jensen V, Walsh JA, Walker PJ, Wang D, Wang LF, Wetzel T, Whitfield AE, Xie JT, Yuen KY, Zhang YZ, Kuhn JH, 2016. Taxonomy of the order Mononegavirales: update 2016. Arch Virol 161, 2351–2360. - PMC - PubMed
-
- Bonavia A, Franti M, Pusateri Keaney E, Kuhen K, Seepersaud M, Radetich B, Shao J, Honda A, Dewhurst J, Balabanis K, Monroe J, Wolff K, Osborne C, Lanieri L, Hoffmaster K, Amin J, Markovits J, Broome M, Skuba E, Cornella-Taracido I, Joberty G, Bouwmeester T, Hamann L, Tallarico JA, Tommasi R, Compton T, Bushell SM, 2011. Identification of broad-spectrum antiviral compounds and assessment of the druggability of their target for efficacy against respiratory syncytial virus (RSV). Proc Natl Acad Sci U S A 108, 6739–6744. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
