

Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen
- PMID: 30150418
- PMCID: PMC6140516
- DOI: 10.1073/pnas.1806002115
Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen
Abstract
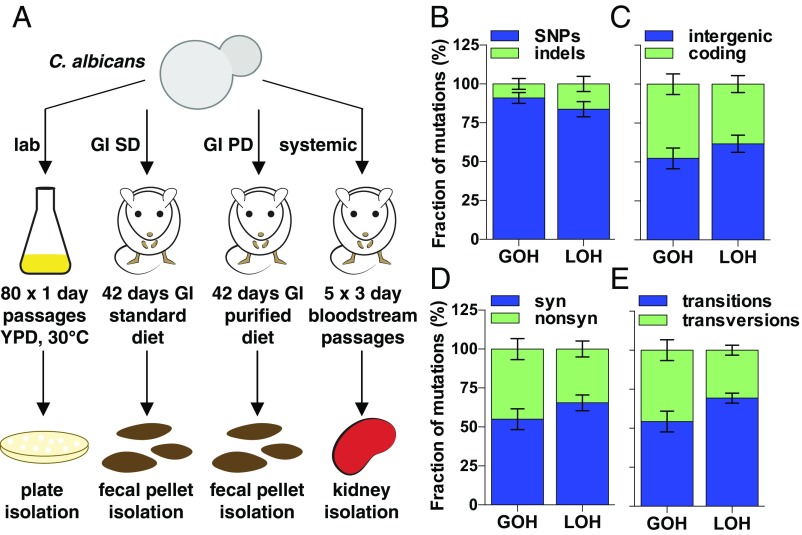
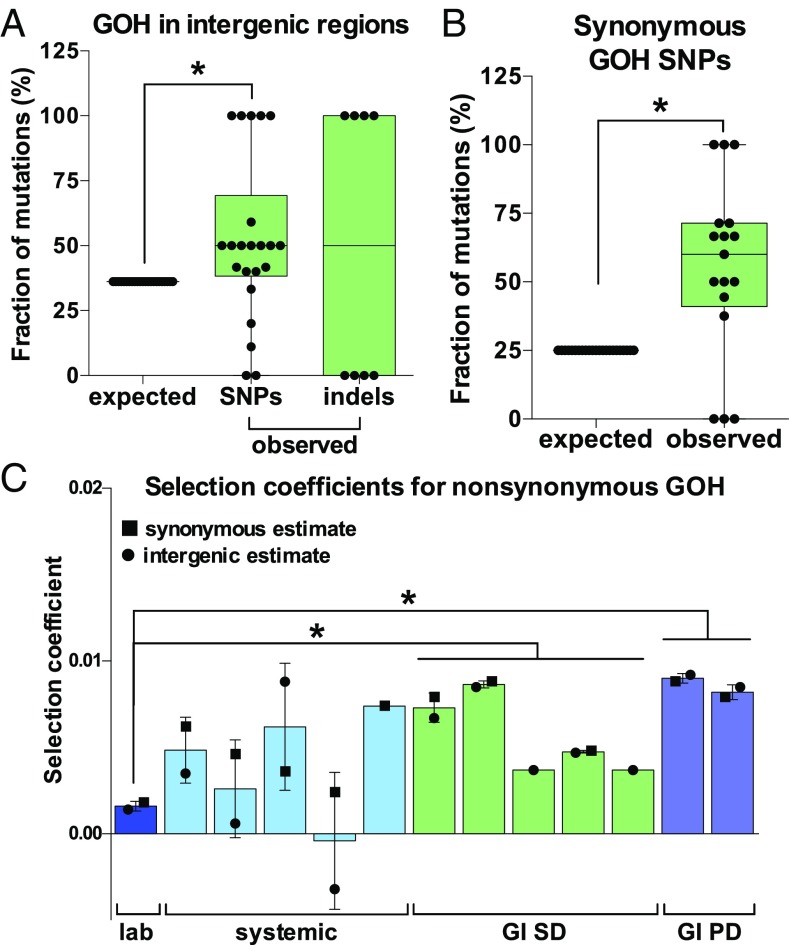
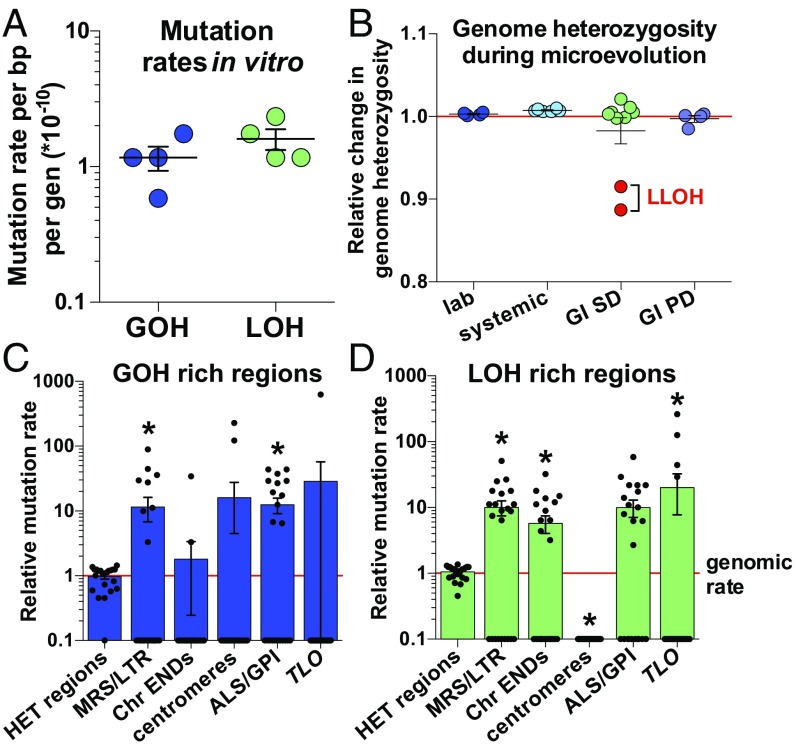
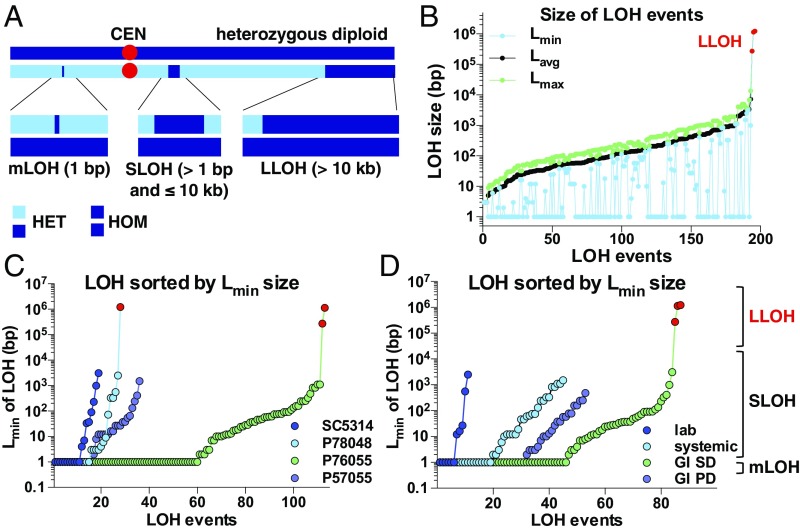
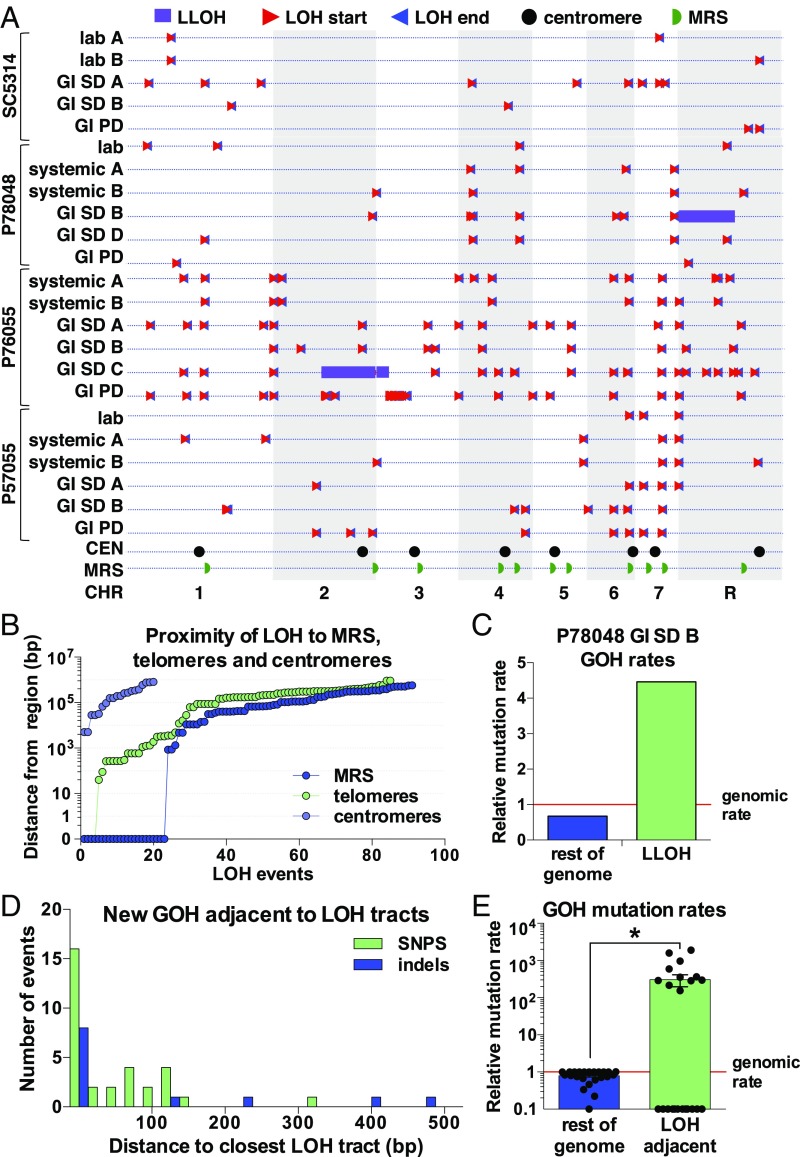
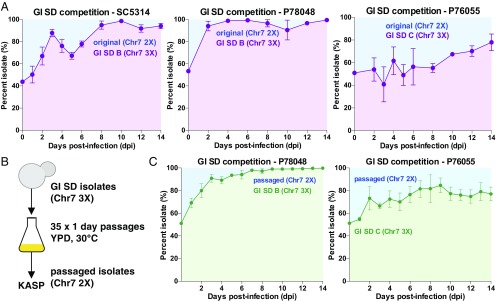
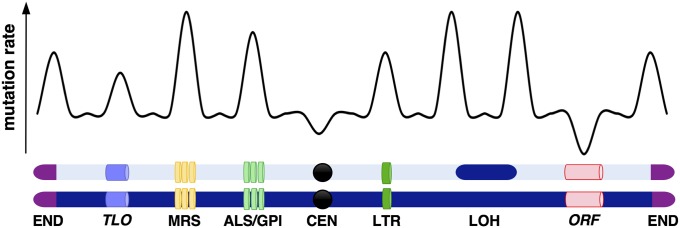
Candida albicans is a heterozygous diploid yeast that is a commensal of the human gastrointestinal tract and a prevalent opportunistic pathogen. Here, whole-genome sequencing was performed on multiple C. albicans isolates passaged both in vitro and in vivo to characterize the complete spectrum of mutations arising in laboratory culture and in the mammalian host. We establish that, independent of culture niche, microevolution is primarily driven by de novo base substitutions and frequent short-tract loss-of-heterozygosity events. An average base-substitution rate of ∼1.2 × 10-10 per base pair per generation was observed in vitro, with higher rates inferred during host infection. Large-scale chromosomal changes were relatively rare, although chromosome 7 trisomies frequently emerged during passaging in a gastrointestinal model and was associated with increased fitness for this niche. Multiple chromosomal features impacted mutational patterns, with mutation rates elevated in repetitive regions, subtelomeric regions, and in gene families encoding cell surface proteins involved in host adhesion. Strikingly, de novo mutation rates were more than 800-fold higher in regions immediately adjacent to emergent loss-of-heterozygosity tracts, indicative of recombination-induced mutagenesis. Furthermore, genomes showed biased patterns of mutations suggestive of extensive purifying selection during passaging. These results reveal how both cell-intrinsic and cell-extrinsic factors influence C. albicans microevolution, and provide a quantitative picture of genome dynamics in this heterozygous diploid species.
Keywords: Candida albicans; LOH; aneuploidy; diploid species; microevolution.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Yang S, et al. Parent-progeny sequencing indicates higher mutation rates in heterozygotes. Nature. 2015;523:463–467. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
