

Impact of Inflammation on Ferritin, Hepcidin and the Management of Iron Deficiency Anemia in Chronic Kidney Disease
- PMID: 30150549
- PMCID: PMC6163440
- DOI: 10.3390/nu10091173
Impact of Inflammation on Ferritin, Hepcidin and the Management of Iron Deficiency Anemia in Chronic Kidney Disease
Abstract
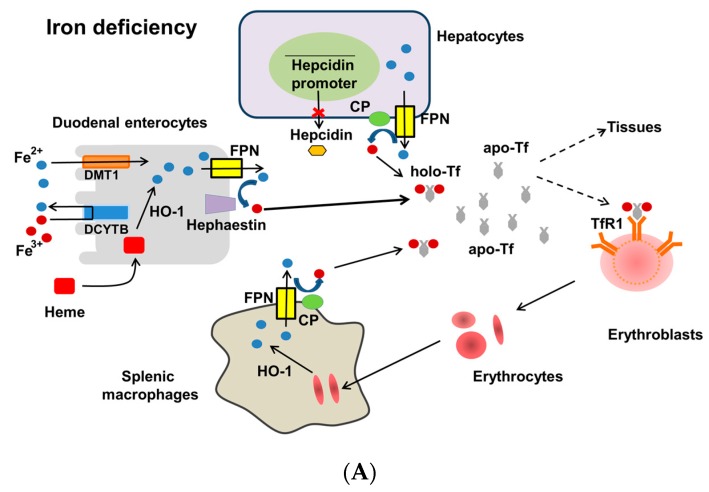
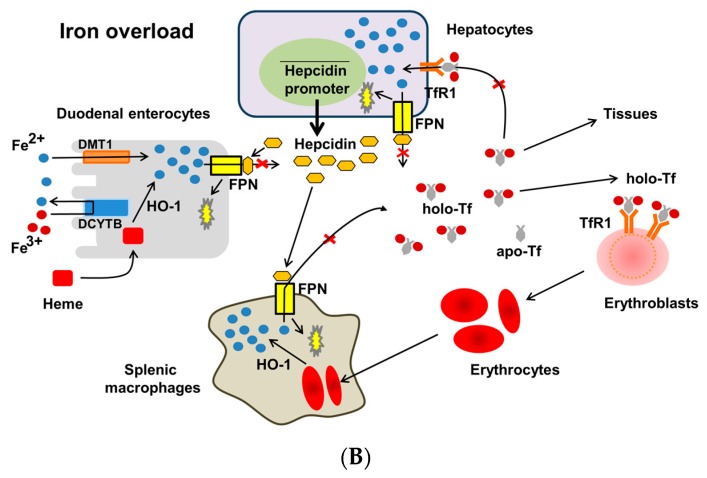
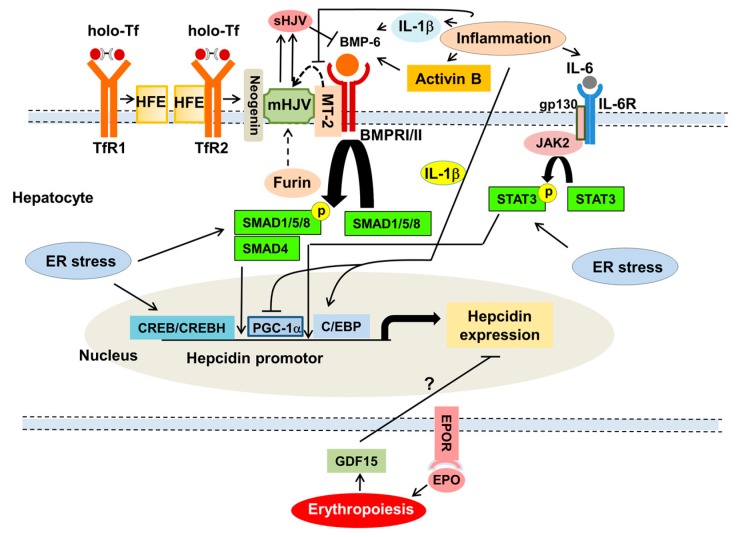
Iron deficiency anemia (IDA) is a major problem in chronic kidney disease (CKD), causing increased mortality. Ferritin stores iron, representing iron status. Hepcidin binds to ferroportin, thereby inhibiting iron absorption/efflux. Inflammation in CKD increases ferritin and hepcidin independent of iron status, which reduce iron availability. While intravenous iron therapy (IIT) is superior to oral iron therapy (OIT) in CKD patients with inflammation, OIT is as effective as IIT in those without. Inflammation reduces predictive values of ferritin and hepcidin for iron status and responsiveness to iron therapy. Upper limit of ferritin to predict iron overload is higher in CKD patients with inflammation than in those without. However, magnetic resonance imaging studies show lower cutoff levels of serum ferritin to predict iron overload in dialysis patients with apparent inflammation than upper limit of ferritin proposed by international guidelines. Compared to CKD patients with inflammation, optimal ferritin levels for IDA are lower in those without, requiring reduced iron dose and leading to decreased mortality. The management of IDA should differ between CKD patients with and without inflammation and include minimization of inflammation. Further studies are needed to determine the impact of inflammation on ferritin, hepcidin and therapeutic strategy for IDA in CKD.
Keywords: C-reactive protein; chronic kidney disease; ferritin; hepcidin; inflammation; iron deficiency anemia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Levi M., Rosselli M., Simonetti M., Brignoli O., Cancian M., Masotti A., Pegoraro V., Cataldo N., Heiman F., Chelo M., et al. Epidemiology of iron deficiency anaemia in four European countries: A population-based study in primary care. Eur. J. Haematol. 2016;97:583–593. doi: 10.1111/ejh.12776. - DOI - PubMed
-
- Iimori S., Naito S., Noda Y., Nishida H., Kihira H., Yui N., Okado T., Sasaki S., Uchida S., Rai T. Anaemia management and mortality risk in newly visiting patients with chronic kidney disease in Japan: The CKD-ROUTE study. Nephrology (Carlton) 2015;20:601–608. doi: 10.1111/nep.12493. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
