

Quantitative Proteomics for Xenopus Embryos II, Data Analysis
- PMID: 30151768
- PMCID: PMC6534117
- DOI: 10.1007/978-1-4939-8784-9_14
Quantitative Proteomics for Xenopus Embryos II, Data Analysis
Abstract
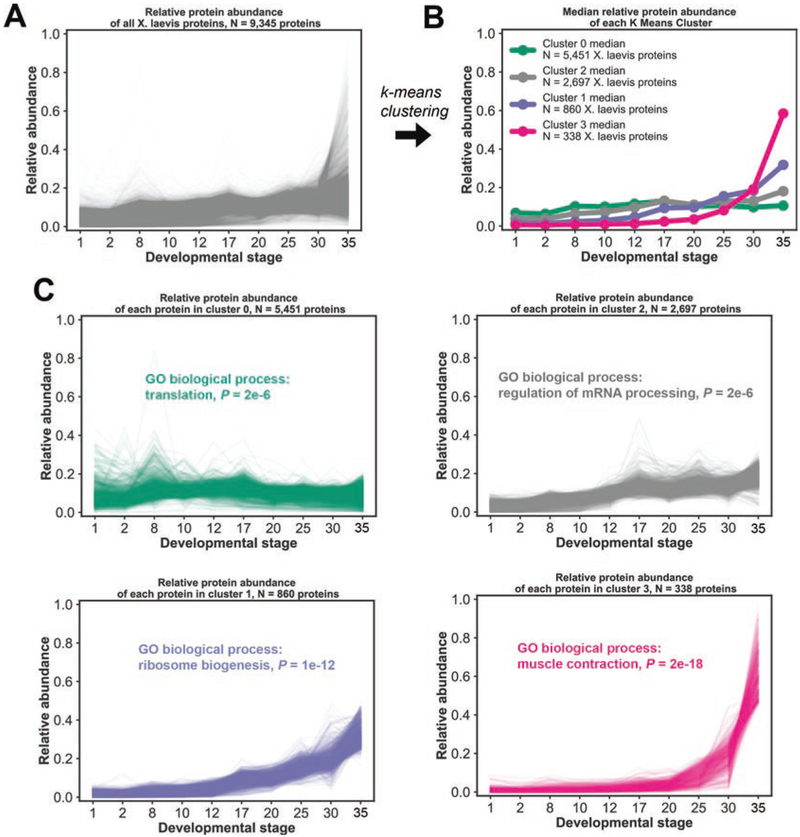
The oocytes, embryos, and cell-free lysates of the frog Xenopus laevis have emerged as powerful models for quantitative proteomic experiments. In the accompanying paper (Chapter 13) we describe how to prepare samples and acquire multiplexed proteomics spectra from those. As an illustrative example we use a 10-stage developmental time series from the egg to stage 35 (just before hatching). Here, we outline how to convert the ~700,000 acquired mass spectra from this time series into protein expression dynamics for ~9000 proteins. We first outline a preliminary quality-control analysis to discover any errors that occurred during sample preparation. We discuss how peptide and protein identification error rates are controlled, and how peptide and protein species are quantified. Our analysis relies on the freely available MaxQuant proteomics pipeline. Finally, we demonstrate how to start interpreting this large dataset by clustering and gene-set enrichment analysis.
Keywords: Development; False discovery rate; Gene symbols; Gene-set enrichment; Mass spectrum; MaxQuant; Quantitative multiplexed proteomics; TMT; Xenopus laevis; k-means clustering.
Figures


References
-
- Gurdon JB, & Wakefield L (1986). Microinjection of amphibian oocytes and eggs for the analysis of transcription. Microinjection and Organelle Transplantation Techniques, 269–299.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
