

Cystic fibrosis transmembrane conductance regulator (CFTR): Making an ion channel out of an active transporter structure
- PMID: 30152709
- PMCID: PMC6986785
- DOI: 10.1080/19336950.2018.1502585
Cystic fibrosis transmembrane conductance regulator (CFTR): Making an ion channel out of an active transporter structure
Abstract
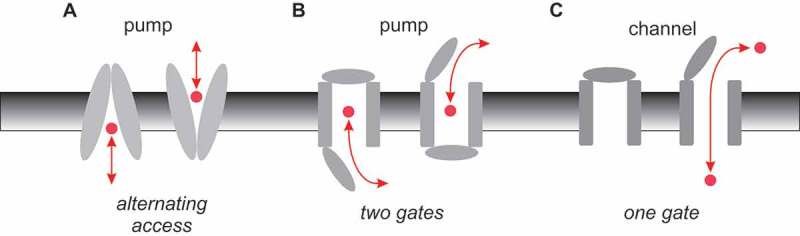
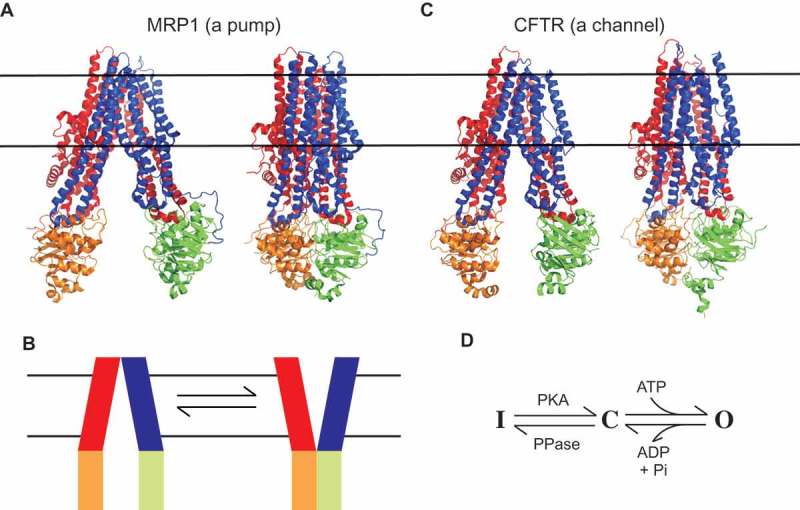
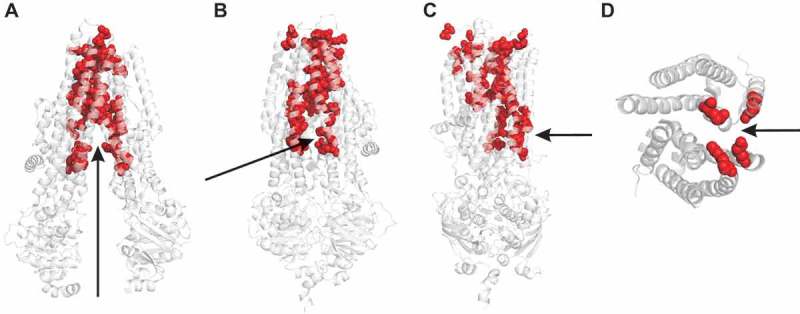
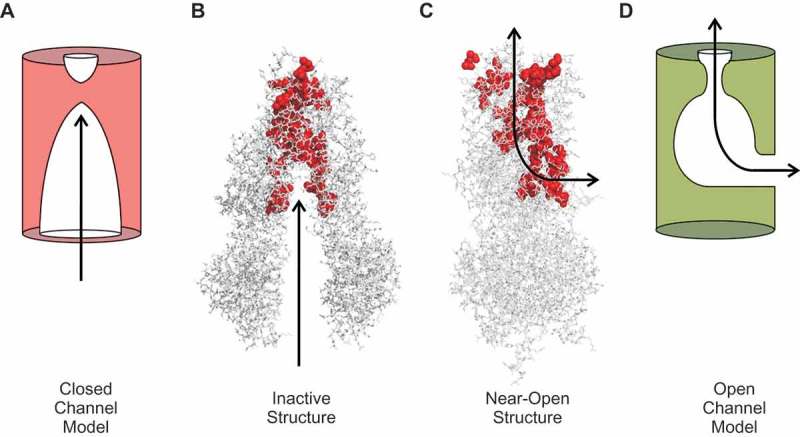
Cystic fibrosis is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR). CFTR is a member of the ATP-binding cassette (ABC) family of membrane transport proteins, most members of which function as ATP-dependent pumps. CFTR is unique among human ABC proteins in functioning not as a pump, but as an ion channel. Recent structural data has indicated that CFTR shares broadly similar overall architecture and ATP-dependent conformational changes as other ABC proteins. Functional investigations suggest that CFTR has a unique open portal connecting the cytoplasm to the transmembrane channel pore, that allows for a continuous pathway for Cl- ions to cross the membrane in one conformation. This lateral portal may be what allows CFTR to function as an ion channel rather than as a pump, suggesting a plausible mechanism by which channel function may have evolved in CFTR.
Keywords: ABC protein; CFTR; channel pore; chloride channel; cystic fibrosis; ion channel.
Figures
References
-
- Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Dean M, Rzhetsky A, Alikmets R.. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. - PubMed
-
- Locher KP. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat Struct Mol Biol. 2016;23:487–493. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical