

Macrophages and Cardiovascular Health
- PMID: 30156496
- PMCID: PMC6442921
- DOI: 10.1152/physrev.00068.2017
Macrophages and Cardiovascular Health
Abstract
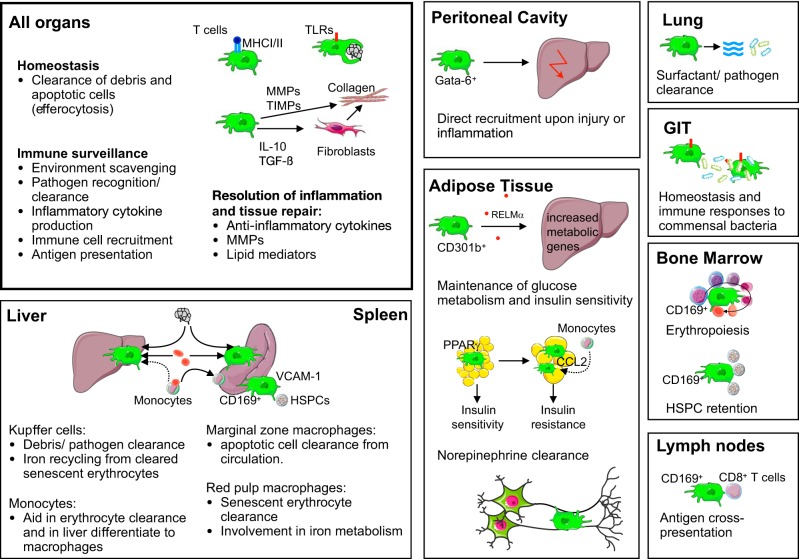
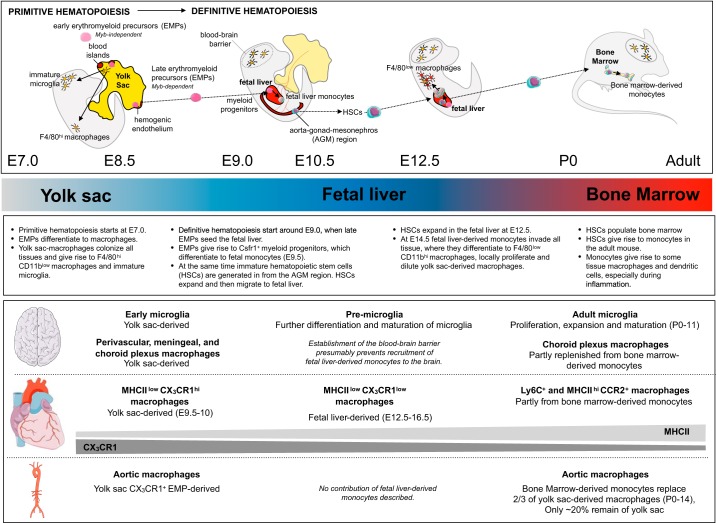
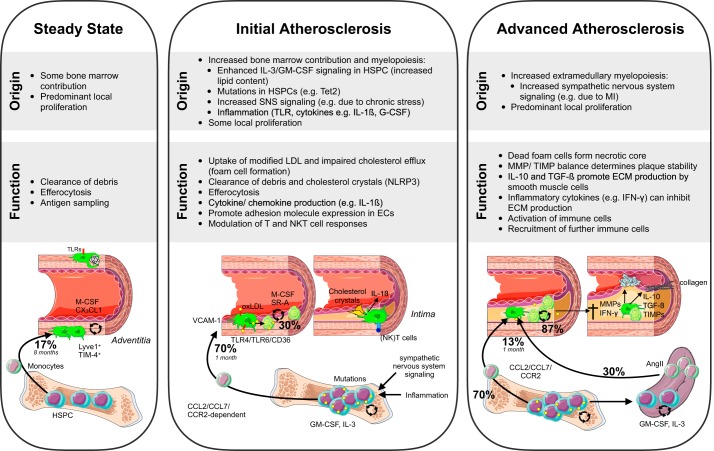
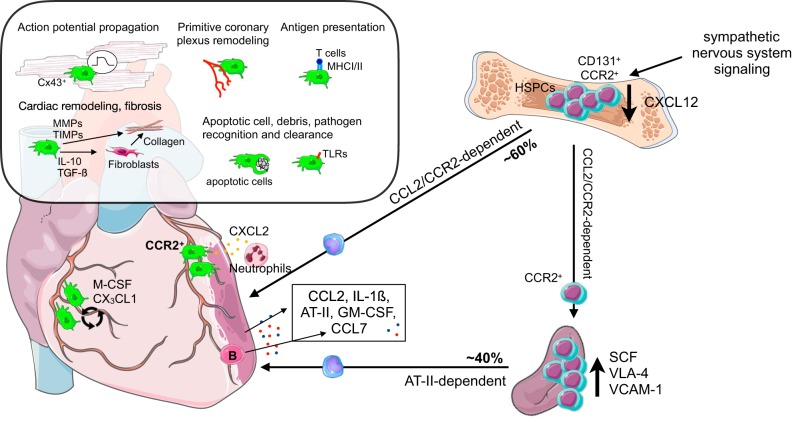
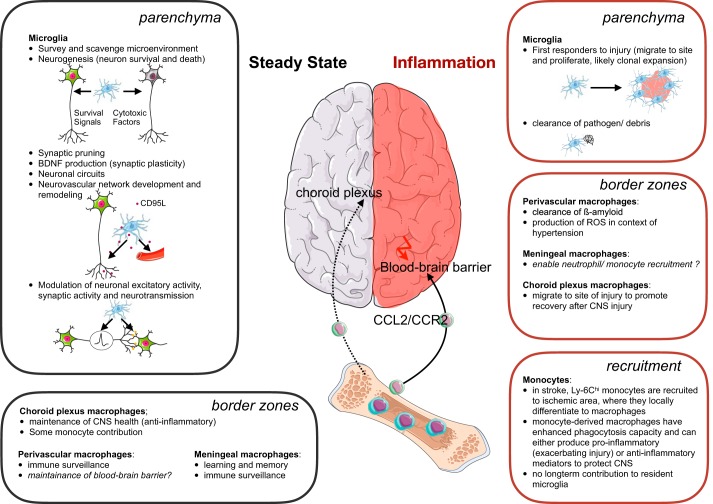
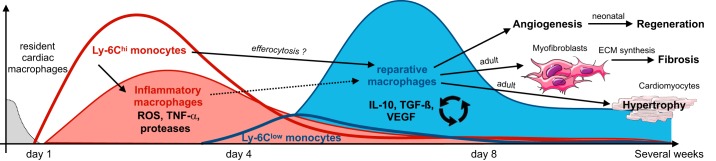
Research during the last decade has generated numerous insights on the presence, phenotype, and function of myeloid cells in cardiovascular organs. Newer tools with improved detection sensitivities revealed sizable populations of tissue-resident macrophages in all major healthy tissues. The heart and blood vessels contain robust numbers of these cells; for instance, 8% of noncardiomyocytes in the heart are macrophages. This number and the cell's phenotype change dramatically in disease conditions. While steady-state macrophages are mostly monocyte independent, macrophages residing in the inflamed vascular wall and the diseased heart derive from hematopoietic organs. In this review, we will highlight signals that regulate macrophage supply and function, imaging applications that can detect changes in cell numbers and phenotype, and opportunities to modulate cardiovascular inflammation by targeting macrophage biology. We strive to provide a systems-wide picture, i.e., to focus not only on cardiovascular organs but also on tissues involved in regulating cell supply and phenotype, as well as comorbidities that promote cardiovascular disease. We will summarize current developments at the intersection of immunology, detection technology, and cardiovascular health.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
-
- A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Díaz M, Gallardo G, Ruiz de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31: 245–258, 2009. doi: 10.1016/j.immuni.2009.06.018. - DOI - PMC - PubMed
-
- Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, Roach LM, Arena RA, Roberts CS, Varma A, Gelwix CC, Salloum FN, Hastillo A, Dinarello CA, Vetrovec GW; VCU-ART Investigators . Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am J Cardiol 105: 1371–1377.e1, 2010. doi: 10.1016/j.amjcard.2009.12.059. - DOI - PubMed
-
- Ait-Oufella H, Kinugawa K, Zoll J, Simon T, Boddaert J, Heeneman S, Blanc-Brude O, Barateau V, Potteaux S, Merval R, Esposito B, Teissier E, Daemen MJ, Lesèche G, Boulanger C, Tedgui A, Mallat Z. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation 115: 2168–2177, 2007. doi: 10.1161/CIRCULATIONAHA.106.662080. - DOI - PubMed
-
- Ait-Oufella H, Pouresmail V, Simon T, Blanc-Brude O, Kinugawa K, Merval R, Offenstadt G, Lesèche G, Cohen PL, Tedgui A, Mallat Z. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol 28: 1429–1431, 2008. doi: 10.1161/ATVBAHA.108.169078. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources