

SP1-induced lncRNA AGAP2-AS1 expression promotes chemoresistance of breast cancer by epigenetic regulation of MyD88
- PMID: 30157918
- PMCID: PMC6114182
- DOI: 10.1186/s13046-018-0875-3
SP1-induced lncRNA AGAP2-AS1 expression promotes chemoresistance of breast cancer by epigenetic regulation of MyD88
Retraction in
-
Retraction Note: SP1-induced lncRNA AGAP2-AS1 expression promotes chemoresistance of breast cancer by epigenetic regulation of MyD88.J Exp Clin Cancer Res. 2023 Jan 11;42(1):16. doi: 10.1186/s13046-023-02596-2. J Exp Clin Cancer Res. 2023. PMID: 36631904 Free PMC article. No abstract available.
Abstract
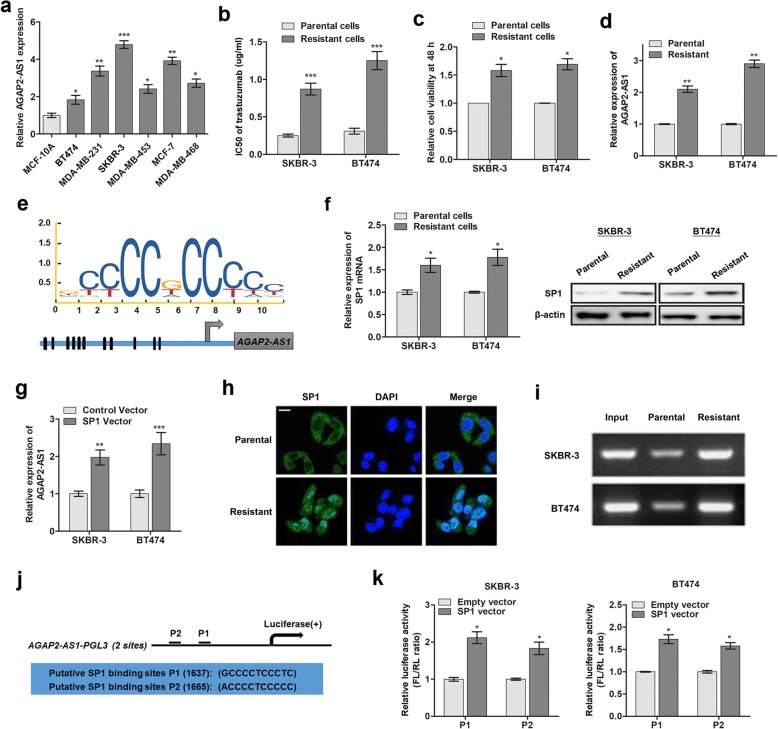
Background: Resistance to trastuzumab has become a leading cause of mortality in breast cancer patients and is one of the major obstacles for improving the clinical outcome. Cell behavior can be modulated by long non-coding RNAs (lncRNAs), but the contribution of lncRNAs in trastuzumab resistance to breast cancer is largely unknown. To this end, the involvement and regulatory function of lncRNA AGAP2-AS1 in human breast cancer are yet to be investigated.
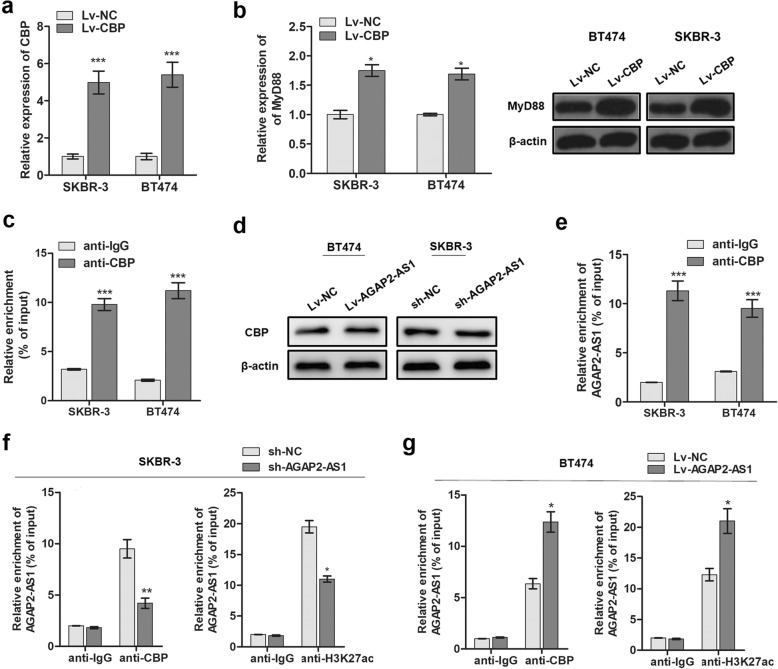
Methods: Trastuzumab-resistant SKBR-3 and BT474 cells were obtained by continuous culture with 5 mg/mL trastuzumab for 6 months. RT-qPCR assay was used to determine the expression of AGAP2-AS1 in tissues and cells. RNA fluorescence in situ hybridization was used to investigate the subcellular location of AGAP2-AS1 in breast cancer cells. Bioinformatic analysis, chromatin immunoprecipitation (ChIP), RNA immunoprecipitation (RIP), western blotting, and immunofluorescence were carried out to verify the regulatory interaction of AGAP2-AS1, CREB-binding protein (CBP), and MyD88. In addition, a series of in vitro assays and a xenograft tumor model were used to analyze the functions of AGAP2-AS1 in breast cancer cells.
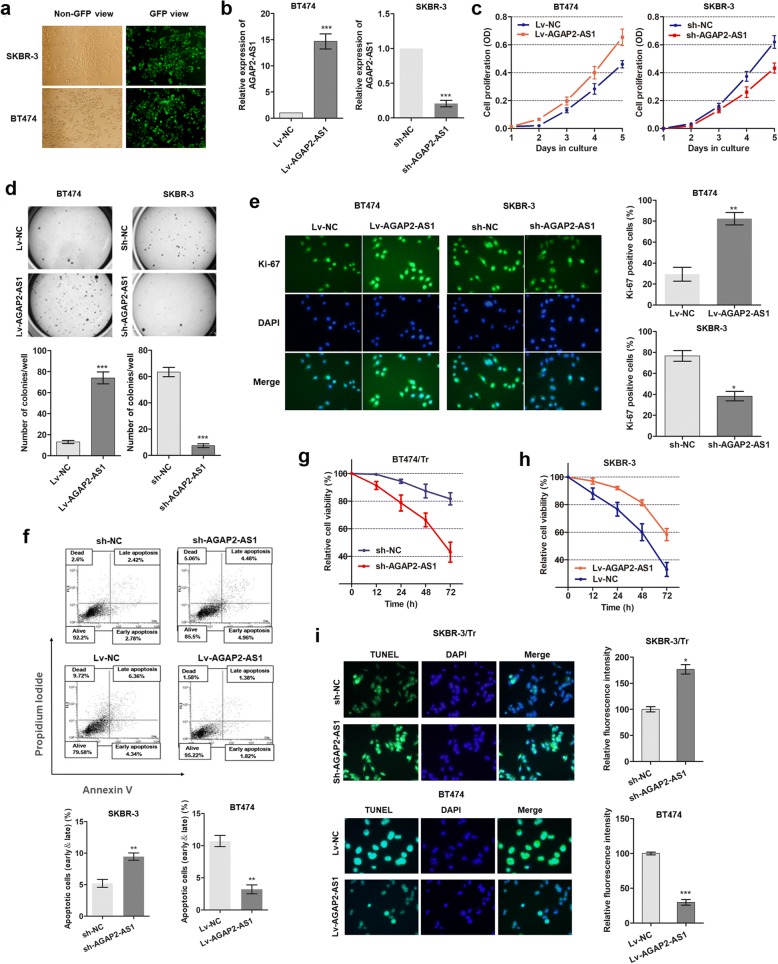
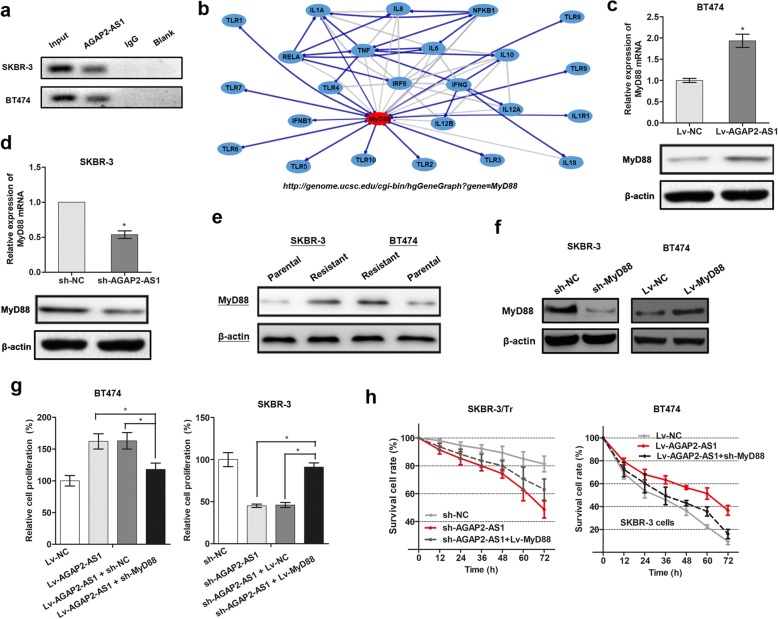
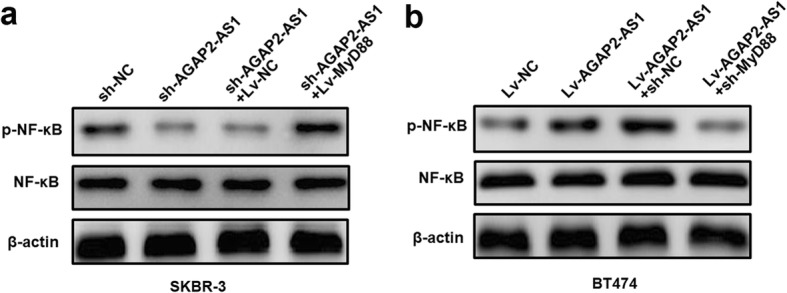
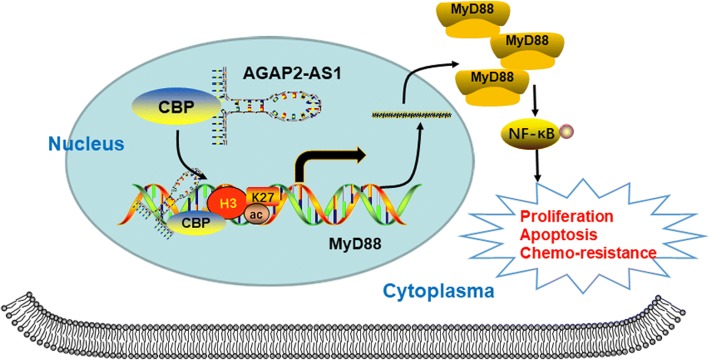
Results: AGAP2-AS1 was upregulated and transcriptionally induced by SP1 in breast cancer. Overexpression of AGAP2-AS1 promoted cell growth, suppressed apoptosis, and caused trastuzumab resistance, whereas knockdown of AGAP2-AS1 showed an opposite effect. MyD88 was identified as a downstream target of AGAP2-AS1 and mediated the AGAP2-AS1-induced oncogenic effects. Mechanistically, the RIP assay revealed that AGAP2-AS1 could bind to CBP, a transcriptional co-activator. ChIP assays showed that AGAP2-AS1-bound CBP increased the enrichment of H3K27ac at the promoter region of MyD88, thus resulting in the upregulation of MyD88. Gain- and loss-of-function assays confirmed that the NF-κB pathway was activated by MyD88 and AGAP2-AS1. Furthermore, high AGAP2-AS1 expression was associated with poor clinical response to trastuzumab therapy in breast cancer patients.
Conclusion: AGAP2-AS1 could promote breast cancer growth and trastuzumab resistance by activating the NF-κB signaling pathway and upregulating MyD88 expression. Therefore, AGAP2-AS1 may serve as a novel biomarker for prognosis and act as a therapeutic target for the trastuzumab treatment.
Keywords: Breast cancer; H3K27 acetylation; MyD88; SP1; Trastuzumab; lncRNA AGAP2-AS1.
Conflict of interest statement
Ethics approval and consent to participate
The present study was authorized by the Ethics Committee of Hainan General Hospital. All procedures performed in studies were in accordance with the ethical standards. All patients and volunteers were anonymous and have provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



References
-
- Robidoux A, Tang G, Rastogi P, Geyer CE, Jr, Azar CA, Atkins JN, Fehrenbacher L, Bear HD, Baez-Diaz L, Sarwar S, et al. Lapatinib as a component of neoadjuvant therapy for HER2-positive operable breast cancer (NSABP protocol B-41): an open-label, randomised phase 3 trial. Lancet Oncol. 2013;14(12):1183–1192. doi: 10.1016/S1470-2045(13)70411-X. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
