

Double genetic disruption of lactate dehydrogenases A and B is required to ablate the "Warburg effect" restricting tumor growth to oxidative metabolism
- PMID: 30158244
- PMCID: PMC6187639
- DOI: 10.1074/jbc.RA118.004180
Double genetic disruption of lactate dehydrogenases A and B is required to ablate the "Warburg effect" restricting tumor growth to oxidative metabolism
Abstract
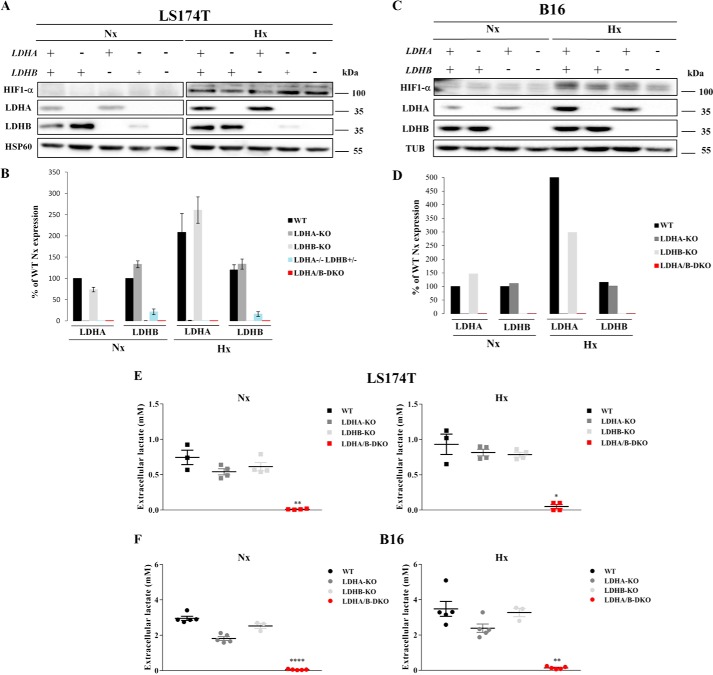
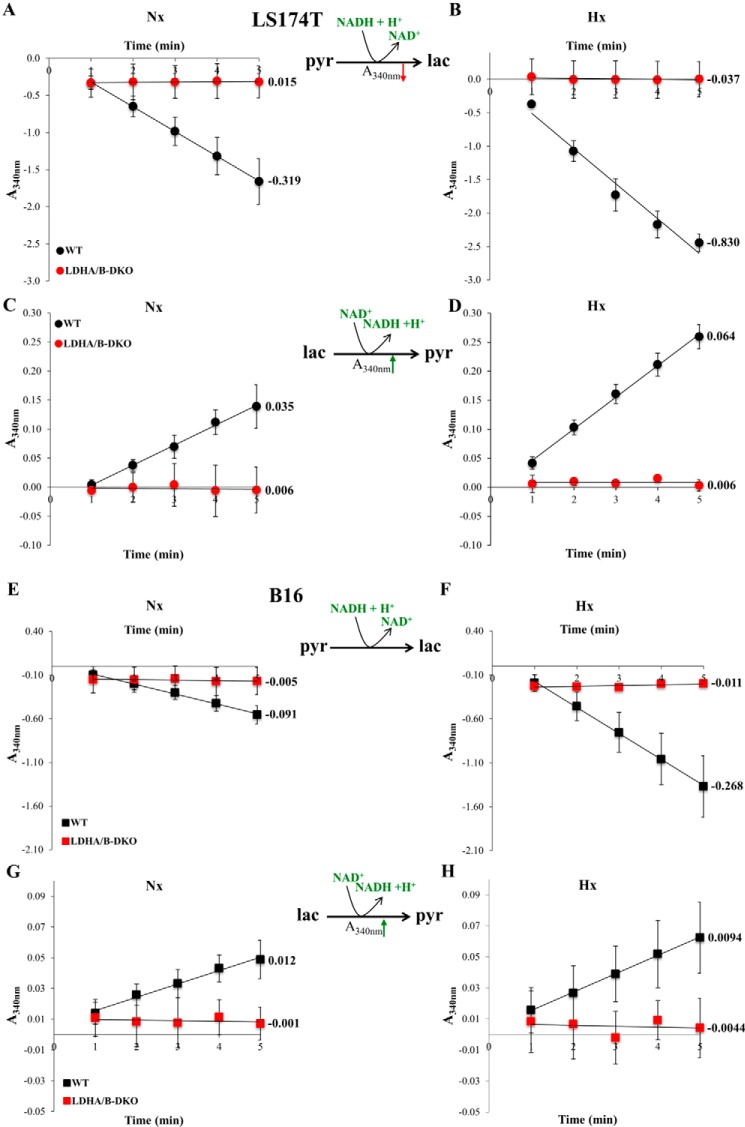
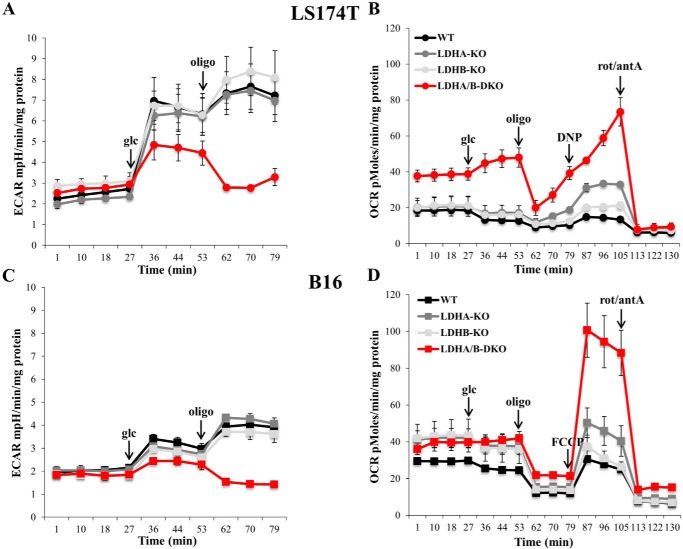
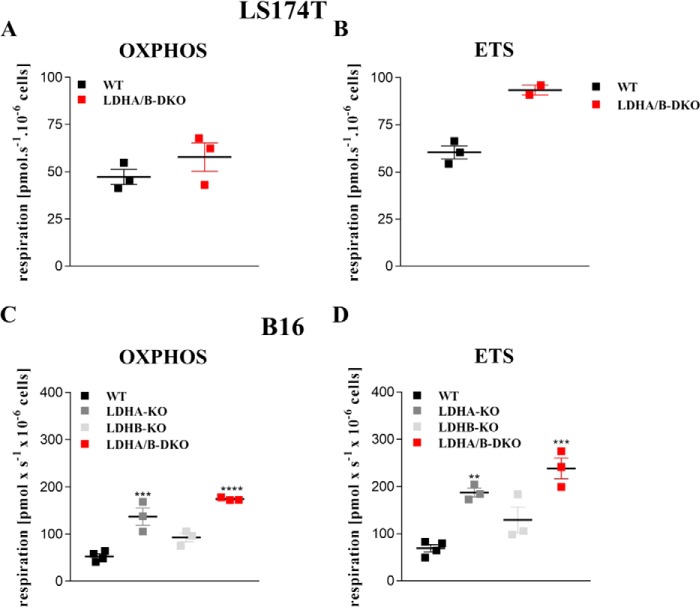
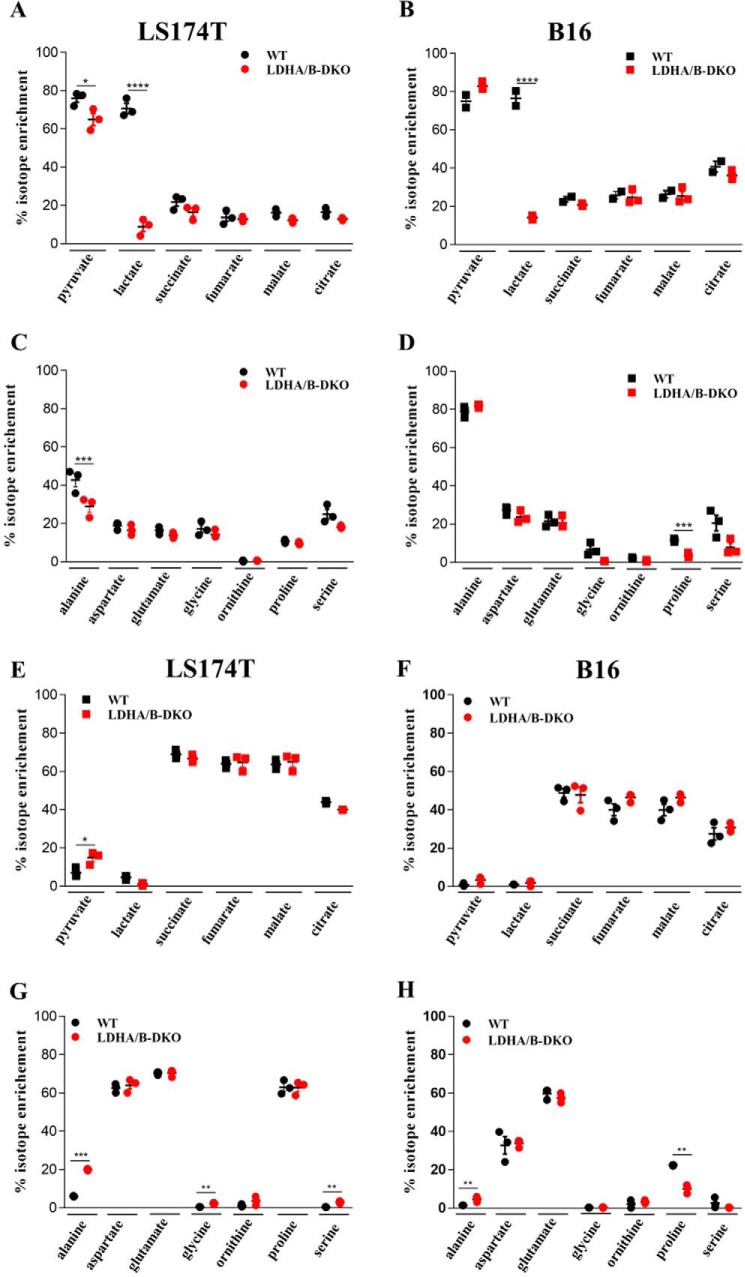
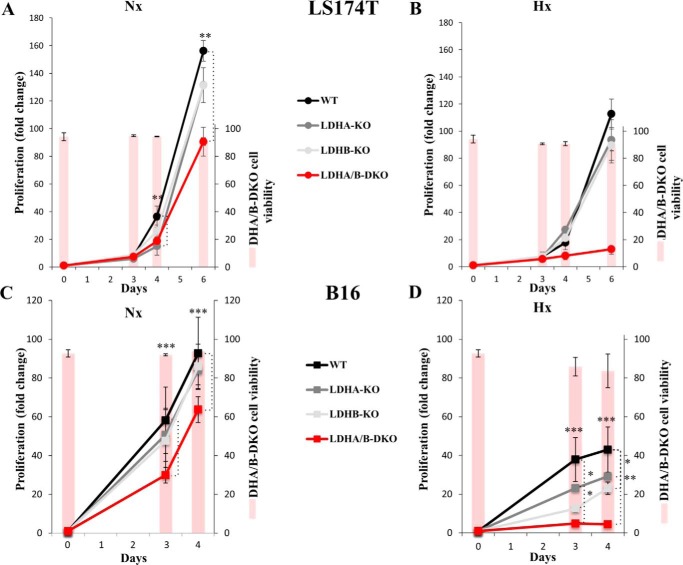
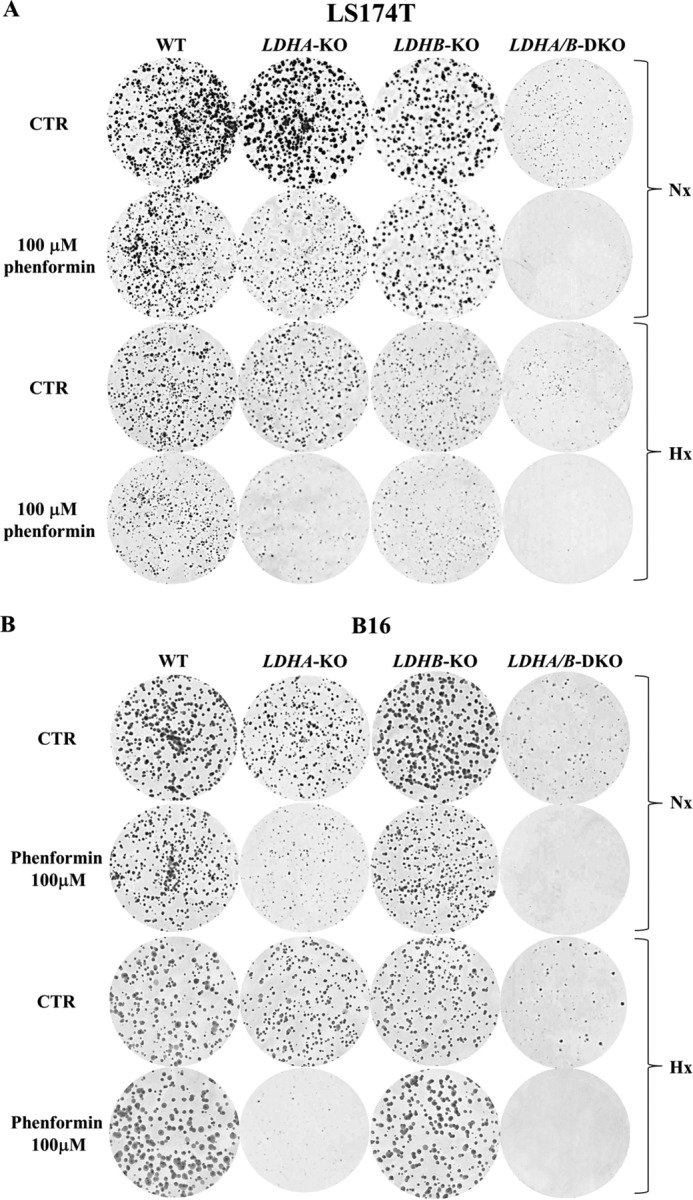
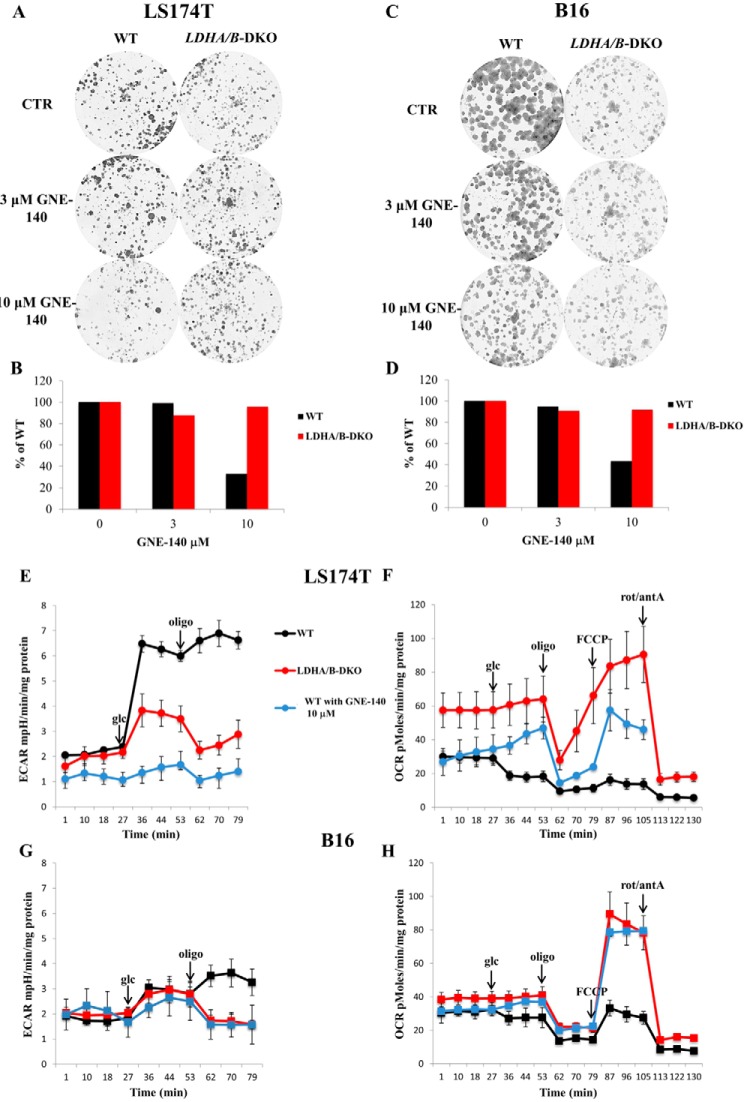
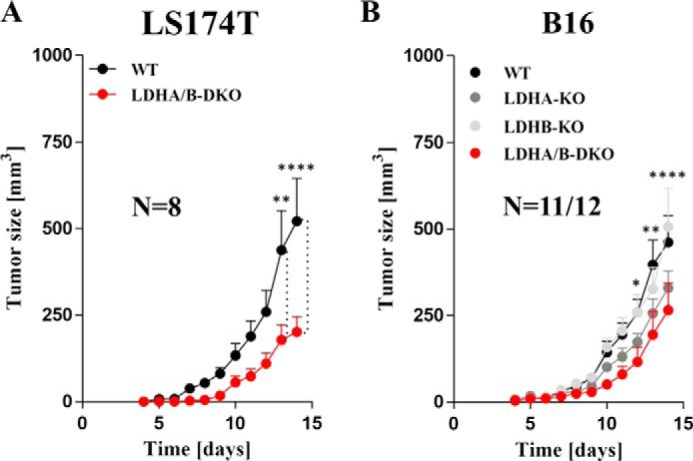
Increased glucose consumption distinguishes cancer cells from normal cells and is known as the "Warburg effect" because of increased glycolysis. Lactate dehydrogenase A (LDHA) is a key glycolytic enzyme, a hallmark of aggressive cancers, and believed to be the major enzyme responsible for pyruvate-to-lactate conversion. To elucidate its role in tumor growth, we disrupted both the LDHA and LDHB genes in two cancer cell lines (human colon adenocarcinoma and murine melanoma cells). Surprisingly, neither LDHA nor LDHB knockout strongly reduced lactate secretion. In contrast, double knockout (LDHA/B-DKO) fully suppressed LDH activity and lactate secretion. Furthermore, under normoxia, LDHA/B-DKO cells survived the genetic block by shifting their metabolism to oxidative phosphorylation (OXPHOS), entailing a 2-fold reduction in proliferation rates in vitro and in vivo compared with their WT counterparts. Under hypoxia (1% oxygen), however, LDHA/B suppression completely abolished in vitro growth, consistent with the reliance on OXPHOS. Interestingly, activation of the respiratory capacity operated by the LDHA/B-DKO genetic block as well as the resilient growth were not consequences of long-term adaptation. They could be reproduced pharmacologically by treating WT cells with an LDHA/B-specific inhibitor (GNE-140). These findings demonstrate that the Warburg effect is not only based on high LDHA expression, as both LDHA and LDHB need to be deleted to suppress fermentative glycolysis. Finally, we demonstrate that the Warburg effect is dispensable even in aggressive tumors and that the metabolic shift to OXPHOS caused by LDHA/B genetic disruptions is responsible for the tumors' escape and growth.
Keywords: CRISPR/Cas; LDHA; LDHB; OXPHOS; Warburg effect; cancer biology; genetic disruption; glucose metabolism; glycolysis; lactate dehydrogenase; lactic acid; metabolic plasticity; pentose phosphate pathway (PPP); tumor growth; tumor metabolism.
© 2018 Ždralević et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article.
Figures
Comment in
-
LDHA and LDHB are dispensable for aerobic glycolysis in neuroblastoma cells while promoting their aggressiveness.J Biol Chem. 2019 Jan 4;294(1):66. doi: 10.1074/jbc.L118.006717. J Biol Chem. 2019. PMID: 30610122 Free PMC article. No abstract available.
-
Reply to Beltinger: Double genetic disruption of lactate dehydrogenases A and B is required to ablate the "Warburg effect" restricting tumor growth to oxidative metabolism.J Biol Chem. 2019 Jan 4;294(1):67. doi: 10.1074/jbc.RL118.006868. J Biol Chem. 2019. PMID: 30610123 Free PMC article. No abstract available.
References
-
- Warburg O. (1956) On respiratory impairment in cancer cells. Science 124, 269–270 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
