

Predicting resistance of clinical Abl mutations to targeted kinase inhibitors using alchemical free-energy calculations
- PMID: 30159405
- PMCID: PMC6110136
- DOI: 10.1038/s42003-018-0075-x
Predicting resistance of clinical Abl mutations to targeted kinase inhibitors using alchemical free-energy calculations
Abstract
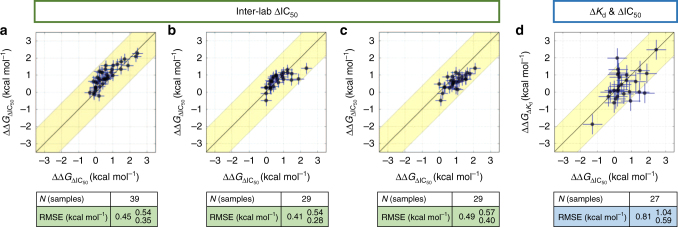
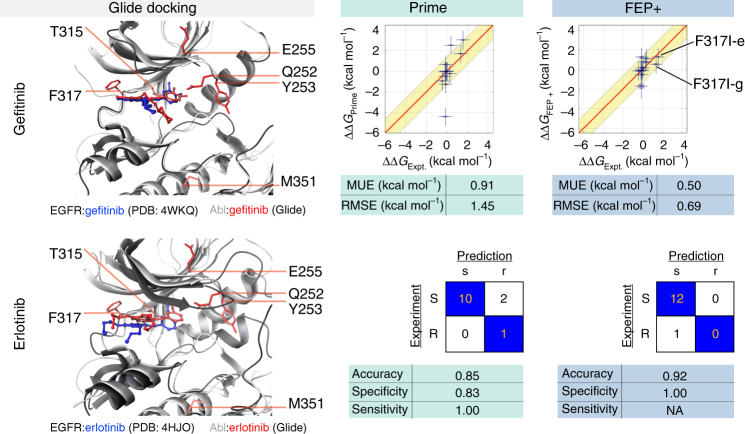
The therapeutic effect of targeted kinase inhibitors can be significantly reduced by intrinsic or acquired resistance mutations that modulate the affinity of the drug for the kinase. In cancer, the majority of missense mutations are rare, making it difficult to predict their impact on inhibitor affinity. This complicates the practice of precision medicine, pairing of patients with clinical trials, and development of next-generation inhibitors. Here, we examine the potential for alchemical free-energy calculations to predict how kinase mutations modulate inhibitor affinities to Abl, a major target in chronic myelogenous leukemia (CML). We find these calculations can achieve useful accuracy in predicting resistance for a set of eight FDA-approved kinase inhibitors across 144 clinically-identified point mutations, achieving a root mean square error in binding free energy changes of kcal/mol (95% confidence interval) and correctly classifying mutations as resistant or susceptible with accuracy. Since these calculations are fast on modern GPUs, this benchmark establishes the potential for physical modeling to collaboratively support the rapid assessment and anticipation of the potential for patient mutations to affect drug potency in clinical applications.
Conflict of interest statement
J.D.C. is a member of the Scientific Advisory Board for Schrödinger Inc. S.R. is a former employee of Schrödinger Inc.; and K.H., C.N., R.A., T.S., and L.W. are employees of Schrödinger Inc.
Figures



References
-
- Roskoski, R. Jr. USFDA approved protein kinase inhibitors. http://www.brimr.org/PKI/PKIs.htm (2017).
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
