

Serum amyloid A - a review
- PMID: 30165816
- PMCID: PMC6117975
- DOI: 10.1186/s10020-018-0047-0
Serum amyloid A - a review
Abstract
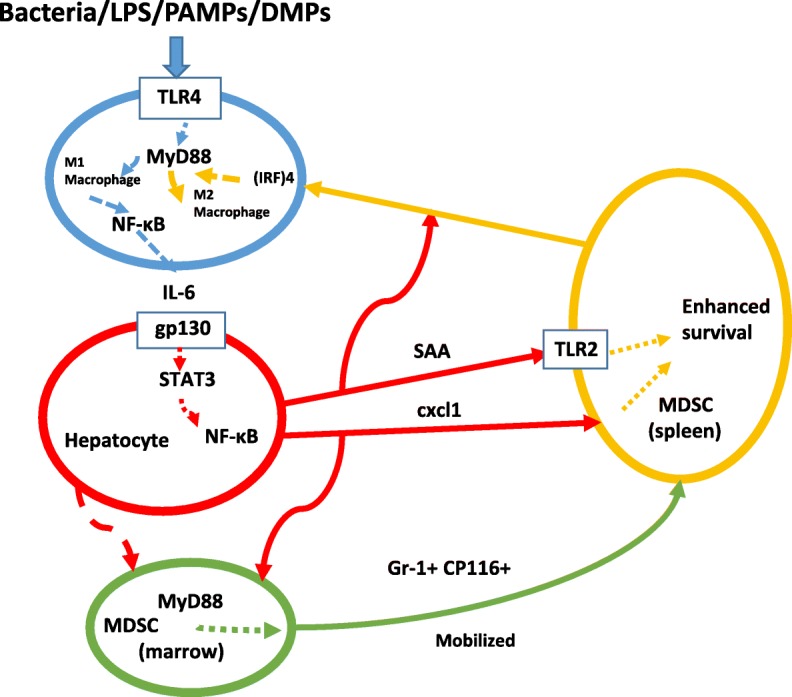
Serum amyloid A (SAA) proteins were isolated and named over 50 years ago. They are small (104 amino acids) and have a striking relationship to the acute phase response with serum levels rising as much as 1000-fold in 24 hours. SAA proteins are encoded in a family of closely-related genes and have been remarkably conserved throughout vertebrate evolution. Amino-terminal fragments of SAA can form highly organized, insoluble fibrils that accumulate in "secondary" amyloid disease. Despite their evolutionary preservation and dynamic synthesis pattern SAA proteins have lacked well-defined physiologic roles. However, considering an array of many, often unrelated, reports now permits a more coordinated perspective. Protein studies have elucidated basic SAA structure and fibril formation. Appreciating SAA's lipophilicity helps relate it to lipid transport and metabolism as well as atherosclerosis. SAA's function as a cytokine-like protein has become recognized in cell-cell communication as well as feedback in inflammatory, immunologic, neoplastic and protective pathways. SAA likely has a critical role in control and possibly propagation of the primordial acute phase response. Appreciating the many cellular and molecular interactions for SAA suggests possibilities for improved understanding of pathophysiology as well as treatment and disease prevention.
Keywords: SAA; Serum amyloid A; acute phase response (APR); amyloidosis; apolipoprotein; arthritis; atherosclerosis; cytokine; inflammation; lipopolysaccharide (LPS); liver; myeloid-derived suppressor cells (MDSC).
Conflict of interest statement
Author’s information
The author received his M.D. and Ph.D. degrees from Johns Hopkins University where he also completed training in Internal Medicine and Medical Genetics. His research has emphasized genetic disorders, amyloid diseases and diagnostic problems in internal medicine. He is a member of the International Society for Amyloidosis, AAAS, ASHG and a founding fellow of ACMG.
Ethics approval
Human subjects were not directly involved in preparing this review
Consent for publication
No individual personal data are involved in this review
Competing interests
The author declares that he has no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



References
-
- Abe-Dohmae S, Kato KH, Kumon Y, Hu W, Ishigami H, Iwamoto N, Okazaki M, Wu C-A, Tsujita M, Ueda K, Yokoyama S. Serum amyloid A generates high density lipoprotein with cellular lipid in an ABCA1- or ABCA7-dependent manner. J Lipid Res. 2006;47:1542–1550. doi: 10.1194/jlr.M600145-JLR200. - DOI - PubMed
-
- Annema W, Nijstad N, Tὄlle M, deBeer JF, Buijs RVC, Heeringa P, van der Giet M, UJF T. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A2. J Lipid Res. 2010;51:743–754. doi: 10.1194/jlr.M000323. - DOI - PMC - PubMed
-
- Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Steoyama H, Nagamori T, Ishikawa E, Shima T, Hara T, Kado S, Jinnohara T, Ohno H, Kondo T, Toyooka K, Watanabe E, Yokoyama S, Tokoro S, Mori H, Noguchi Y, Morita H, Ivanov II, Sugiyama T, Nunez G, Camp JG, Hattori M, Umesaki Y, Honda K. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. 2015;163:357–380. doi: 10.1016/j.cell.2015.08.058. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
