

Comprehensive antibiotic-linked mutation assessment by resistance mutation sequencing (RM-seq)
- PMID: 30165908
- PMCID: PMC6117896
- DOI: 10.1186/s13073-018-0572-z
Comprehensive antibiotic-linked mutation assessment by resistance mutation sequencing (RM-seq)
Abstract
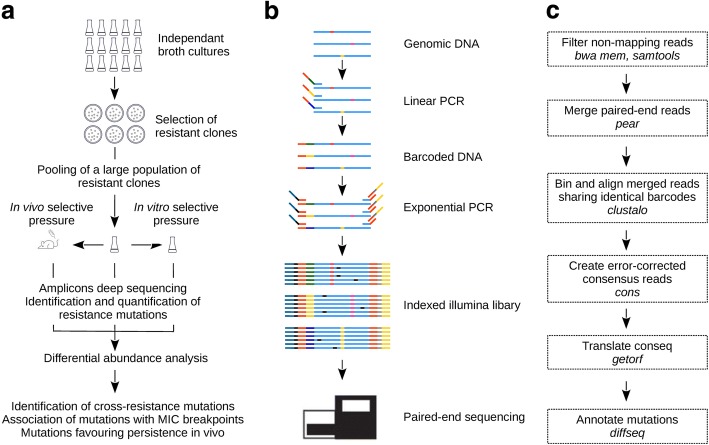
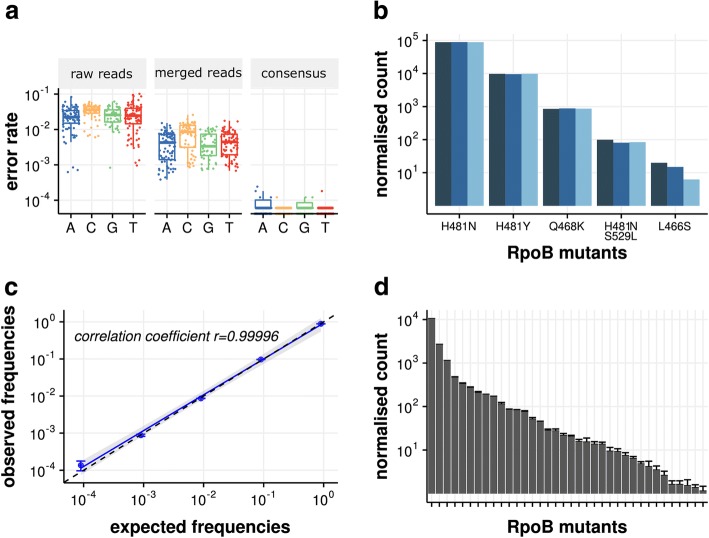
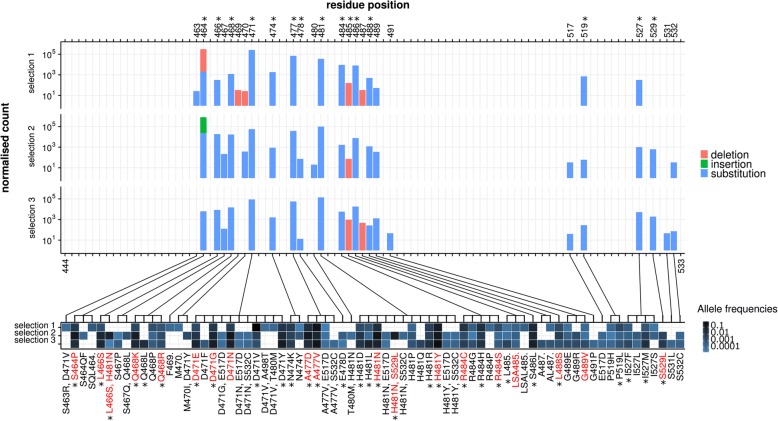
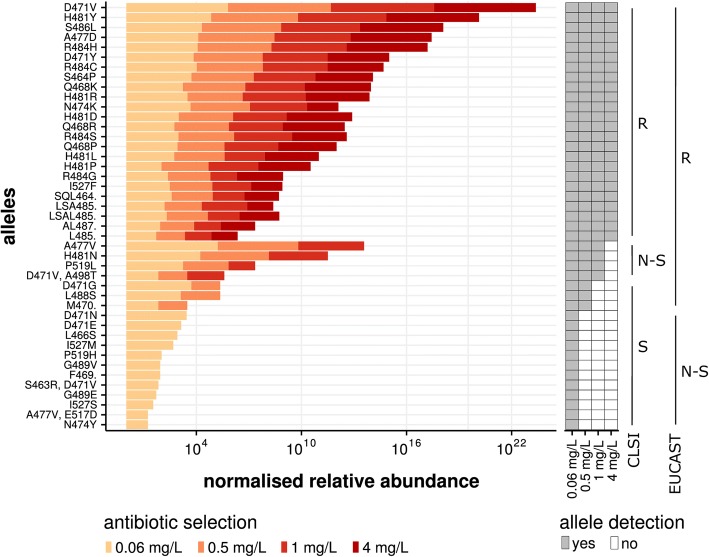
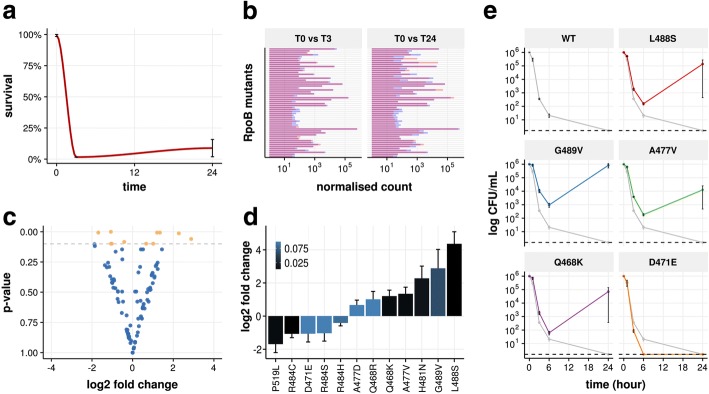
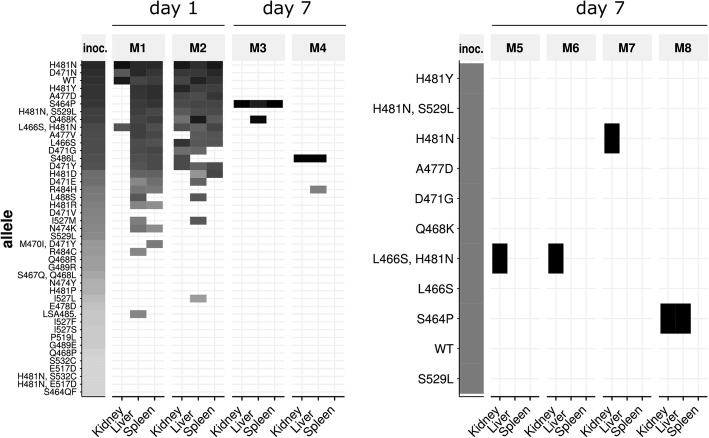
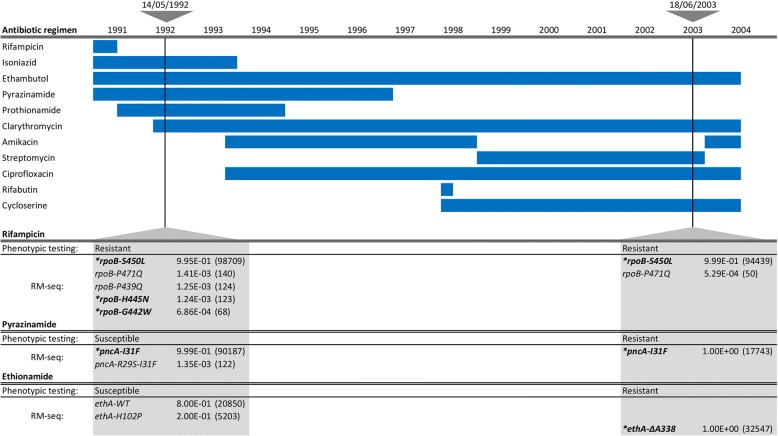
Mutation acquisition is a major mechanism of bacterial antibiotic resistance that remains insufficiently characterised. Here we present RM-seq, a new amplicon-based deep sequencing workflow based on a molecular barcoding technique adapted from Low Error Amplicon sequencing (LEA-seq). RM-seq allows detection and functional assessment of mutational resistance at high throughput from mixed bacterial populations. The sensitive detection of very low-frequency resistant sub-populations permits characterisation of antibiotic-linked mutational repertoires in vitro and detection of rare resistant populations during infections. Accurate quantification of resistance mutations enables phenotypic screening of mutations conferring pleiotropic phenotypes such as in vivo persistence, collateral sensitivity or cross-resistance. RM-seq will facilitate comprehensive detection, characterisation and surveillance of resistant bacterial populations ( https://github.com/rguerillot/RM-seq ).
Keywords: Antibiotic resistance; Daptomycin; Deep sequencing; Mycobacterium tuberculosis; Resistance mutations; Rifampicin; Staphylococcus aureus.
Conflict of interest statement
Ethics approval and consent to participate
Samples from a TB patient were collected as part of the Victorian Department of Health and Human Services TB Control Programme, under the Public Health and Wellbeing Act 2008 (
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- World Health Organization. Antimicrobial resistance: global report on surveillance 2014: World Health Organization; 2014. http://www.who.int/drugresistance/documents/surveillancereport/en/. Accessed 27 July 2018.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
