

Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors
- PMID: 30166462
- PMCID: PMC6176908
- DOI: 10.1126/science.aam8419
Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors
Abstract
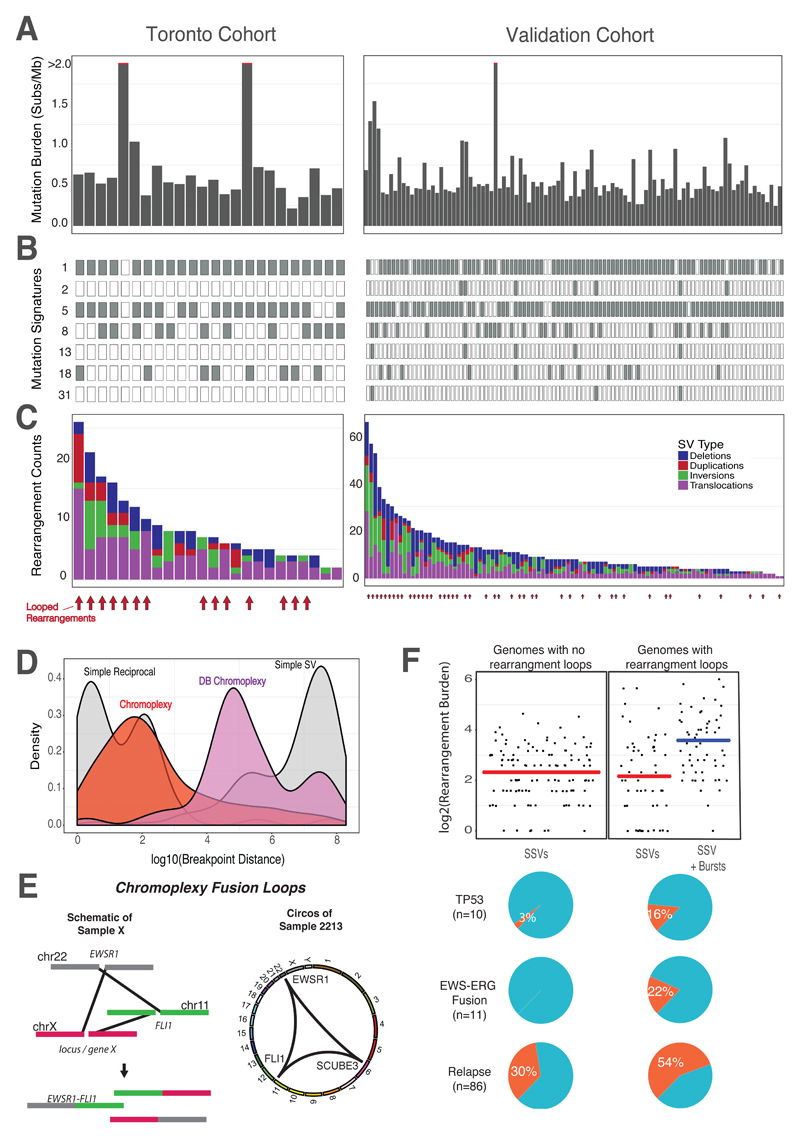
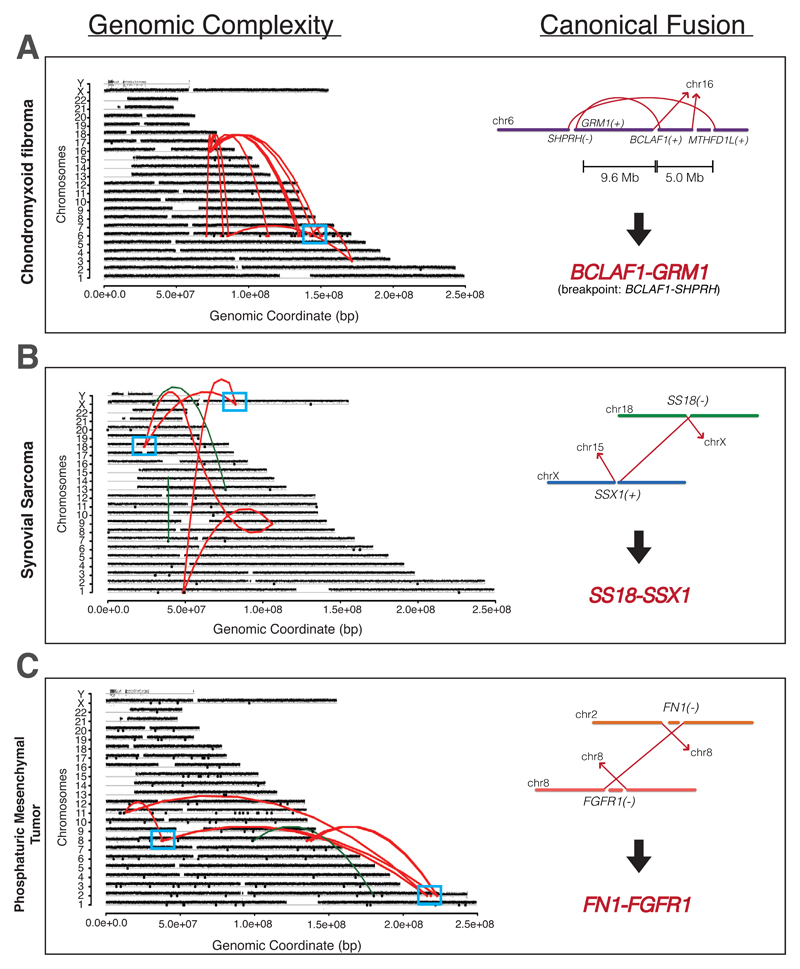
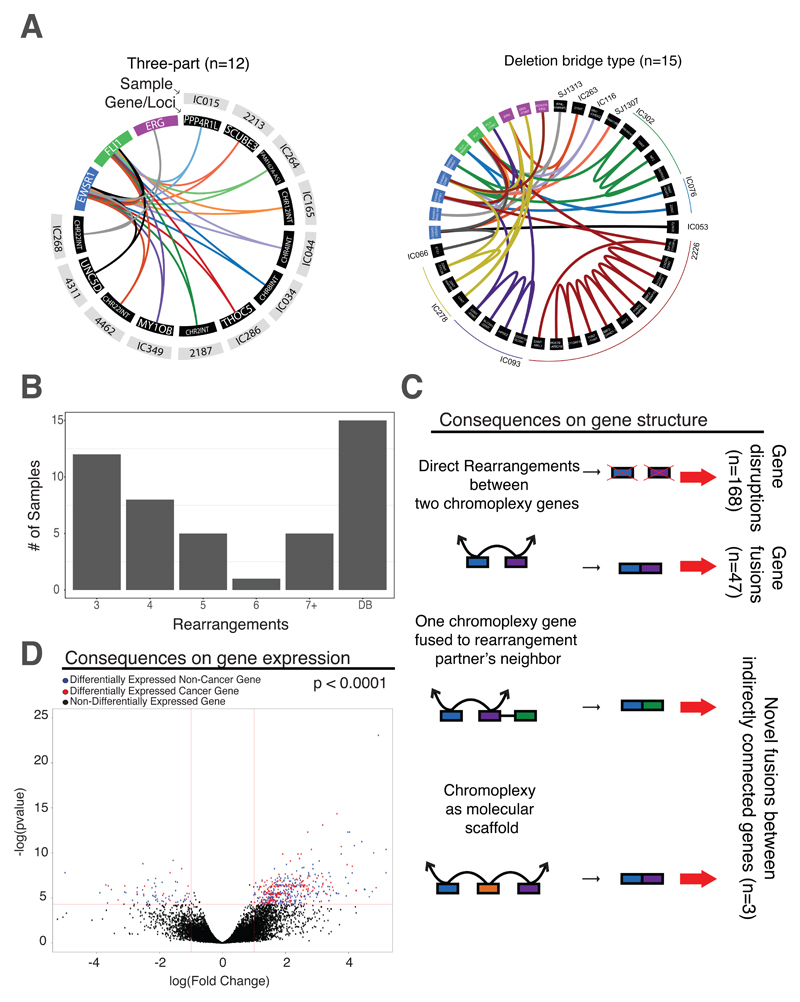
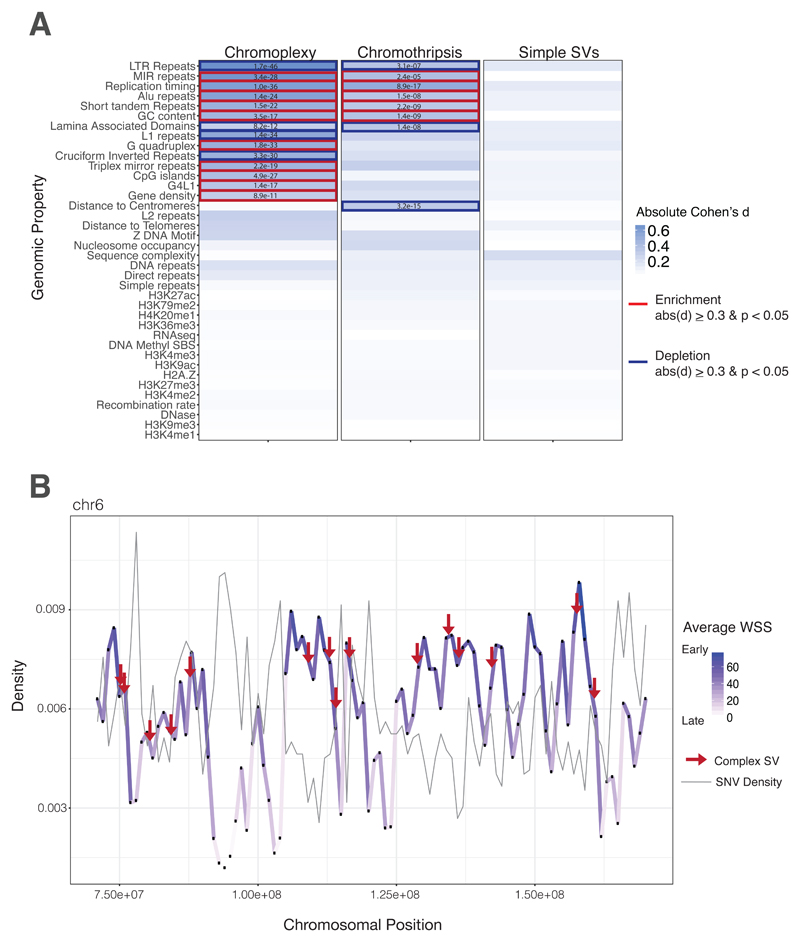
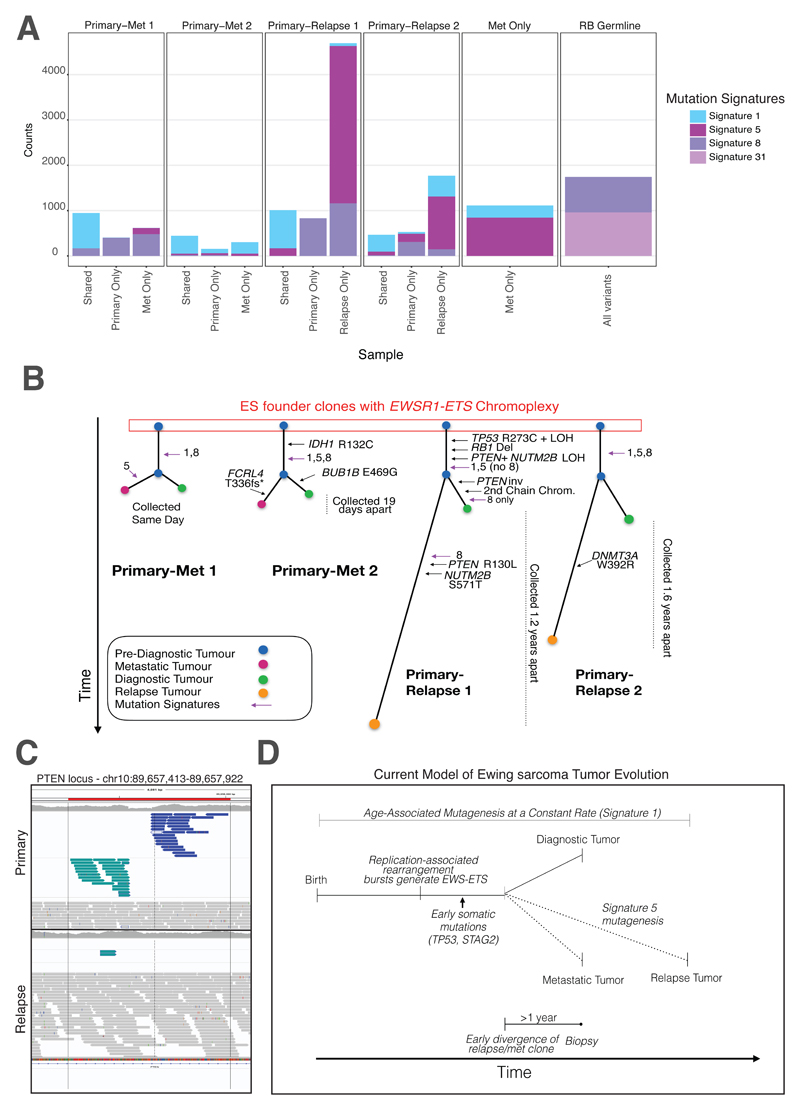
Sarcomas are cancers of the bone and soft tissue often defined by gene fusions. Ewing sarcoma involves fusions between EWSR1, a gene encoding an RNA binding protein, and E26 transformation-specific (ETS) transcription factors. We explored how and when EWSR1-ETS fusions arise by studying the whole genomes of Ewing sarcomas. In 52 of 124 (42%) of tumors, the fusion gene arises by a sudden burst of complex, loop-like rearrangements, a process called chromoplexy, rather than by simple reciprocal translocations. These loops always contained the disease-defining fusion at the center, but they disrupted multiple additional genes. The loops occurred preferentially in early replicating and transcriptionally active genomic regions. Similar loops forming canonical fusions were found in three other sarcoma types. Chromoplexy-generated fusions appear to be associated with an aggressive form of Ewing sarcoma. These loops arise early, giving rise to both primary and relapse Ewing sarcoma tumors, which can continue to evolve in parallel.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Fusion oncogenes-genetic musical chairs.Science. 2018 Aug 31;361(6405):848-849. doi: 10.1126/science.aau8231. Science. 2018. PMID: 30166475 No abstract available.
References
-
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
