

Innate immune activity as a predictor of persistent insulin secretion and association with responsiveness to CTLA4-Ig treatment in recent-onset type 1 diabetes
- PMID: 30167736
- PMCID: PMC6182660
- DOI: 10.1007/s00125-018-4708-x
Innate immune activity as a predictor of persistent insulin secretion and association with responsiveness to CTLA4-Ig treatment in recent-onset type 1 diabetes
Abstract
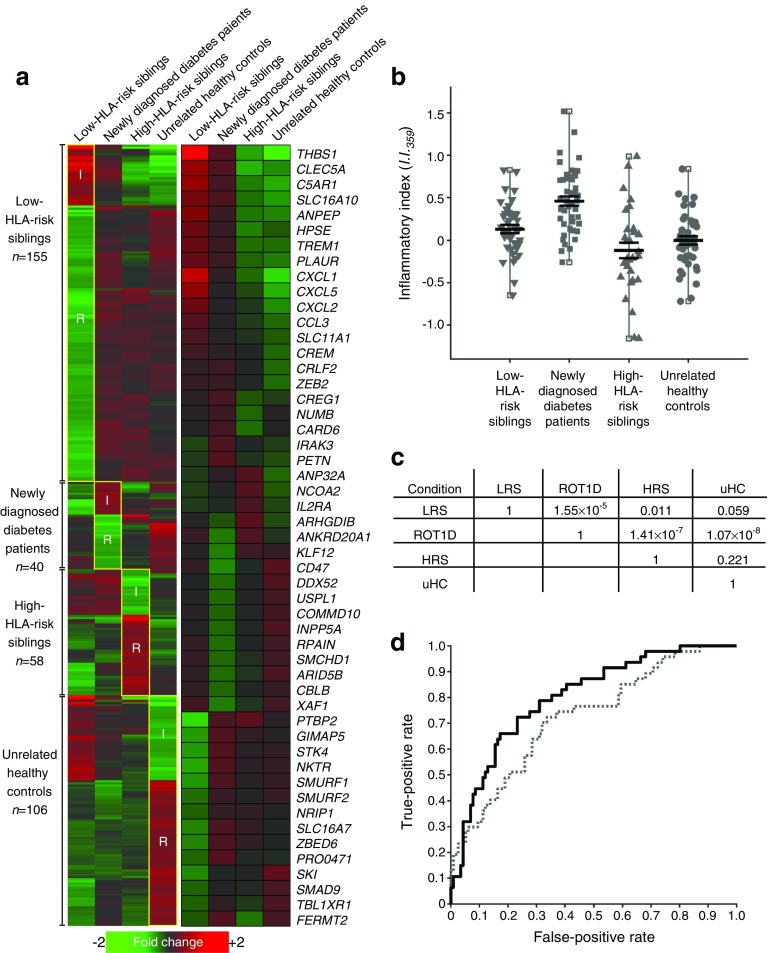
Aims/hypothesis: The study aimed to determine whether discrete subtypes of type 1 diabetes exist, based on immunoregulatory profiles at clinical onset, as this has significant implications for disease treatment and prevention as well as the design and analysis of clinical trials.
Methods: Using a plasma-based transcriptional bioassay and a gene-ontology-based scoring algorithm, we examined local participants from the Children's Hospital of Wisconsin and conducted an ancillary analysis of TrialNet CTLA4-Ig trial (TN-09) participants.
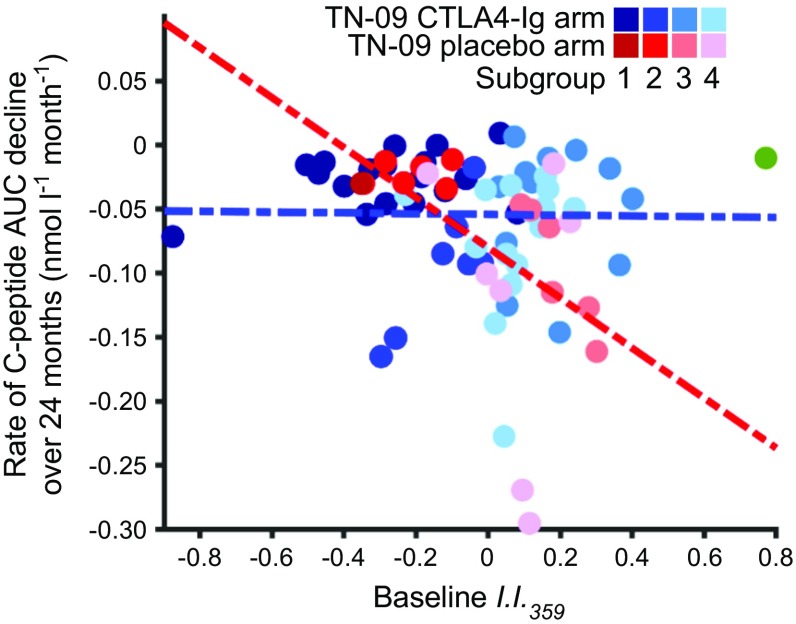
Results: The inflammatory/regulatory balance measured during the post-onset period was highly variable. Notably, a significant inverse relationship was identified between baseline innate inflammatory activity and stimulated C-peptide AUC measured at 3, 6, 12, 18 and 24 months post onset among placebo-treated individuals (p ≤ 0.015). Further, duration of persistent insulin secretion was negatively related to baseline inflammation (p ≤ 0.012) and positively associated with baseline abundance of circulating activated regulatory T cells (CD4+/CD45RA-/FOXP3high; p = 0.016). Based on these findings, data from participants treated with CTLA4-Ig were stratified by inflammatory activity at onset; in this way, we identified pathways and transcripts consistent with inhibition of T cell activation and enhanced immunoregulation. Variance among baseline plasma-induced signatures of TN-09 participants was further examined with weighted gene co-expression network analysis and related to clinical metrics. Four age-independent subgroups were identified that differed in terms of baseline innate inflammatory/regulatory bias, rate of C-peptide decline and response to CTLA4-Ig treatment.
Conclusions/interpretation: These data support the existence of multiple type 1 diabetes subtypes characterised by varying levels of baseline innate inflammation that are associated with the rate of C-peptide decline.
Data availability: Gene expression data files are publicly available through the National Center for Biotechnology Information Gene Expression Omnibus (accession number GSE102234).
Keywords: Biomarker; CTLA4-Ig; Disease heterogeneity; Honeymoon; Partial remission; Therapeutic response; Type 1 diabetes.
Conflict of interest statement
The authors declare that there is no duality of interest associated with this manuscript.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
