

Umap and Bismap: quantifying genome and methylome mappability
- PMID: 30169659
- PMCID: PMC6237805
- DOI: 10.1093/nar/gky677
Umap and Bismap: quantifying genome and methylome mappability
Abstract
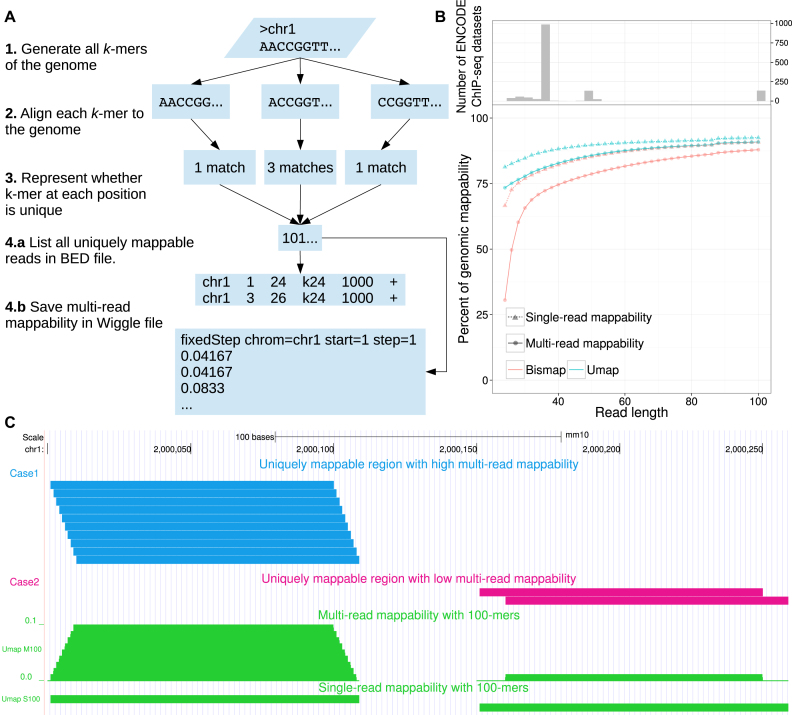
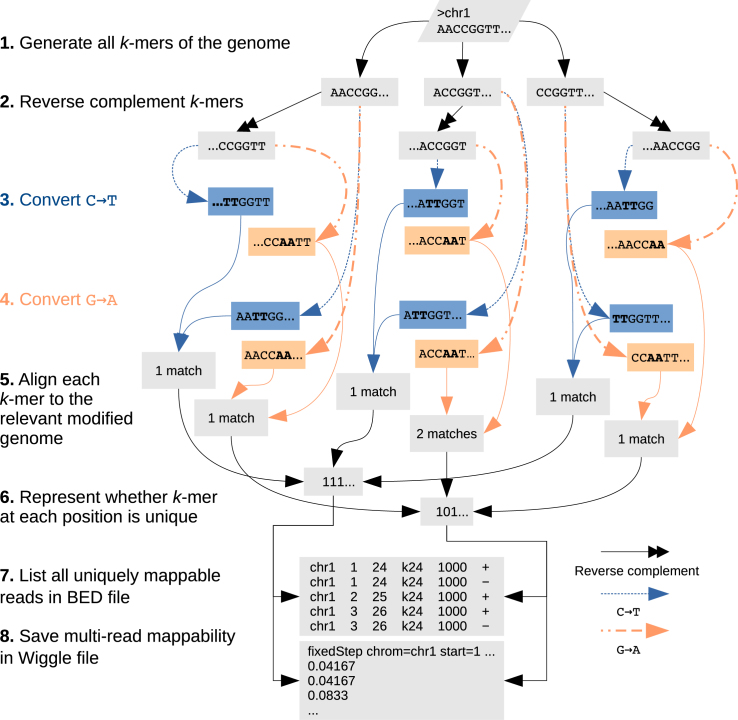
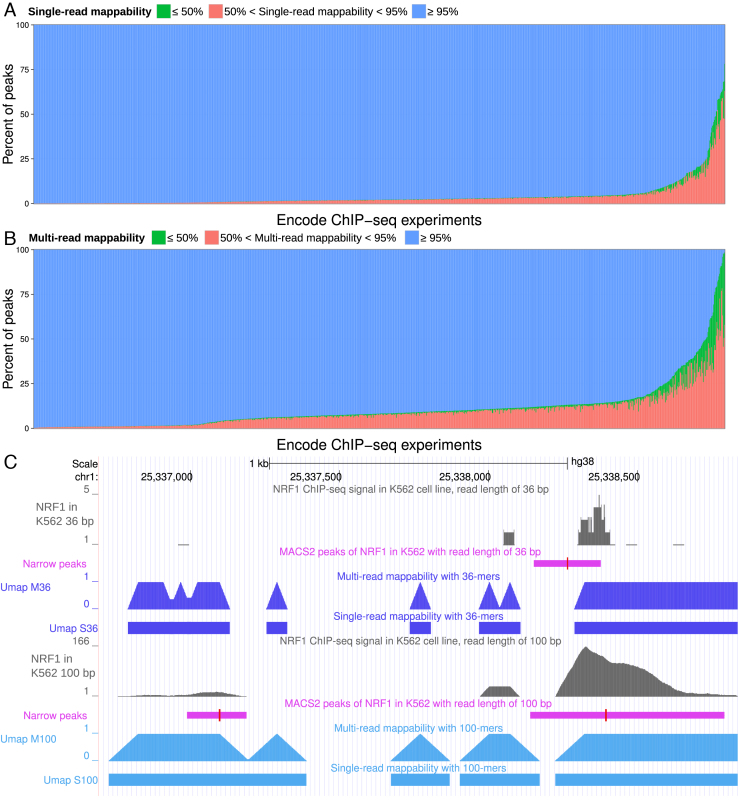
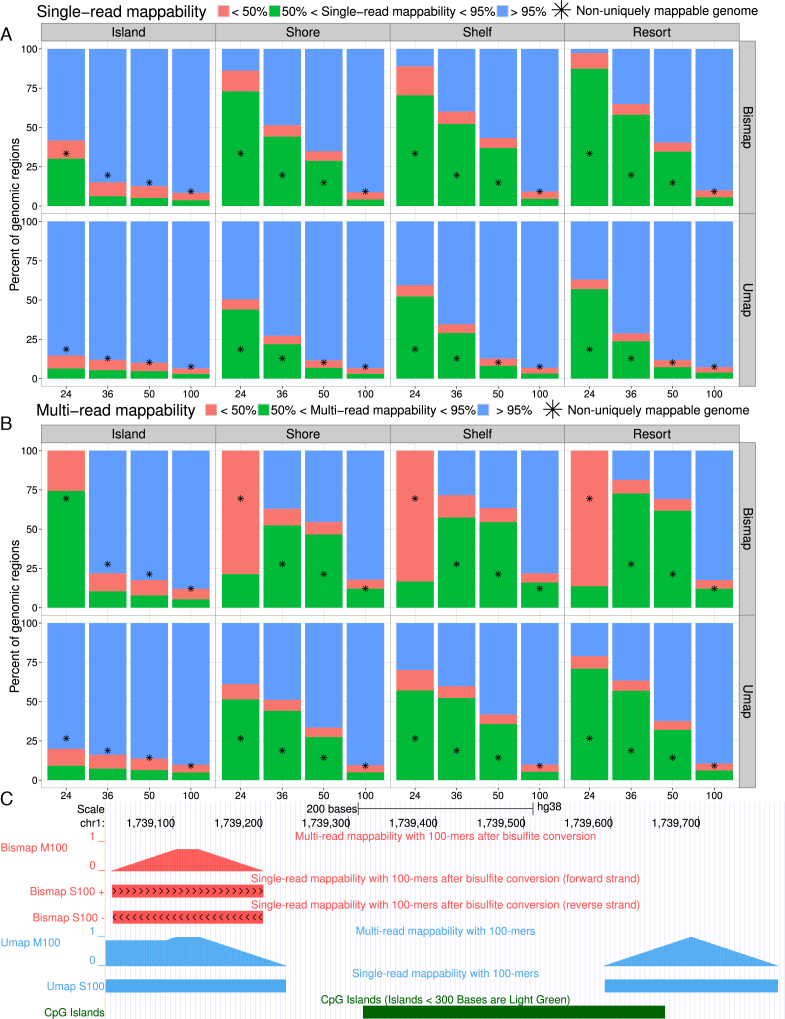
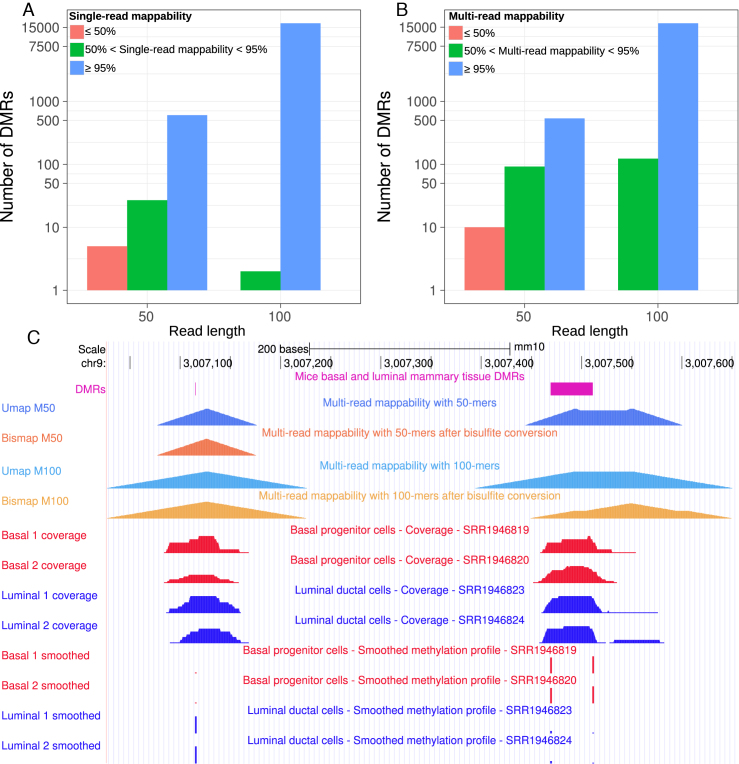
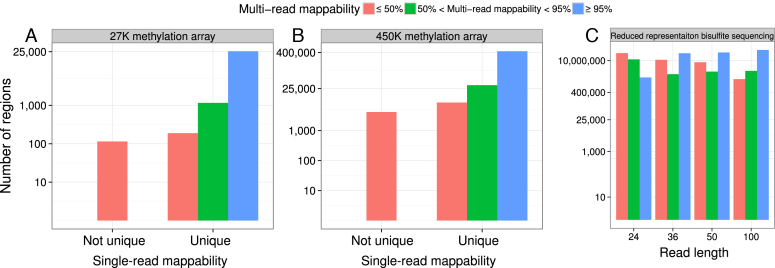
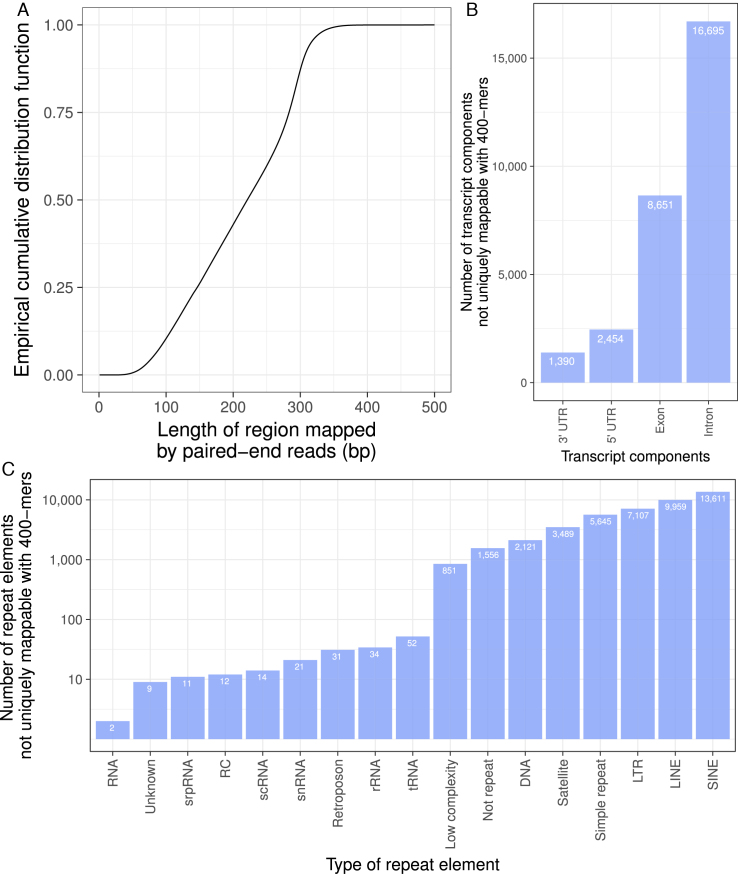
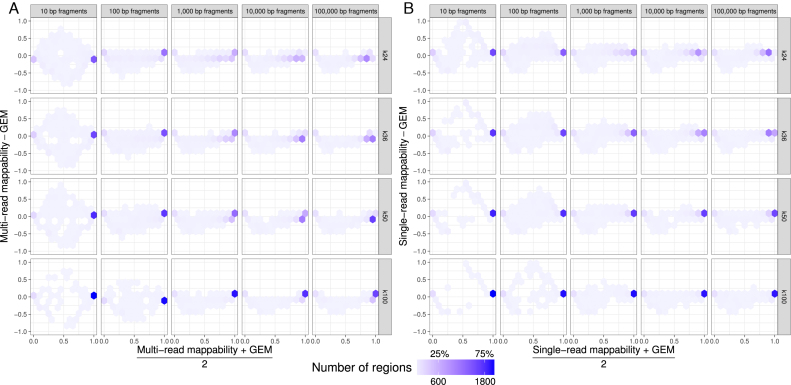
Short-read sequencing enables assessment of genetic and biochemical traits of individual genomic regions, such as the location of genetic variation, protein binding and chemical modifications. Every region in a genome assembly has a property called 'mappability', which measures the extent to which it can be uniquely mapped by sequence reads. In regions of lower mappability, estimates of genomic and epigenomic characteristics from sequencing assays are less reliable. These regions have increased susceptibility to spurious mapping from reads from other regions of the genome with sequencing errors or unexpected genetic variation. Bisulfite sequencing approaches used to identify DNA methylation exacerbate these problems by introducing large numbers of reads that map to multiple regions. Both to correct assumptions of uniformity in downstream analysis and to identify regions where the analysis is less reliable, it is necessary to know the mappability of both ordinary and bisulfite-converted genomes. We introduce the Umap software for identifying uniquely mappable regions of any genome. Its Bismap extension identifies mappability of the bisulfite-converted genome. A Umap and Bismap track hub for human genome assemblies GRCh37/hg19 and GRCh38/hg38, and mouse assemblies GRCm37/mm9 and GRCm38/mm10 is available at https://bismap.hoffmanlab.org for use with genome browsers.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
