

Adenosine Receptor-Mediated Developmental Loss of Spike Timing-Dependent Depression in the Hippocampus
- PMID: 30169759
- PMCID: PMC6644873
- DOI: 10.1093/cercor/bhy194
Adenosine Receptor-Mediated Developmental Loss of Spike Timing-Dependent Depression in the Hippocampus
Abstract
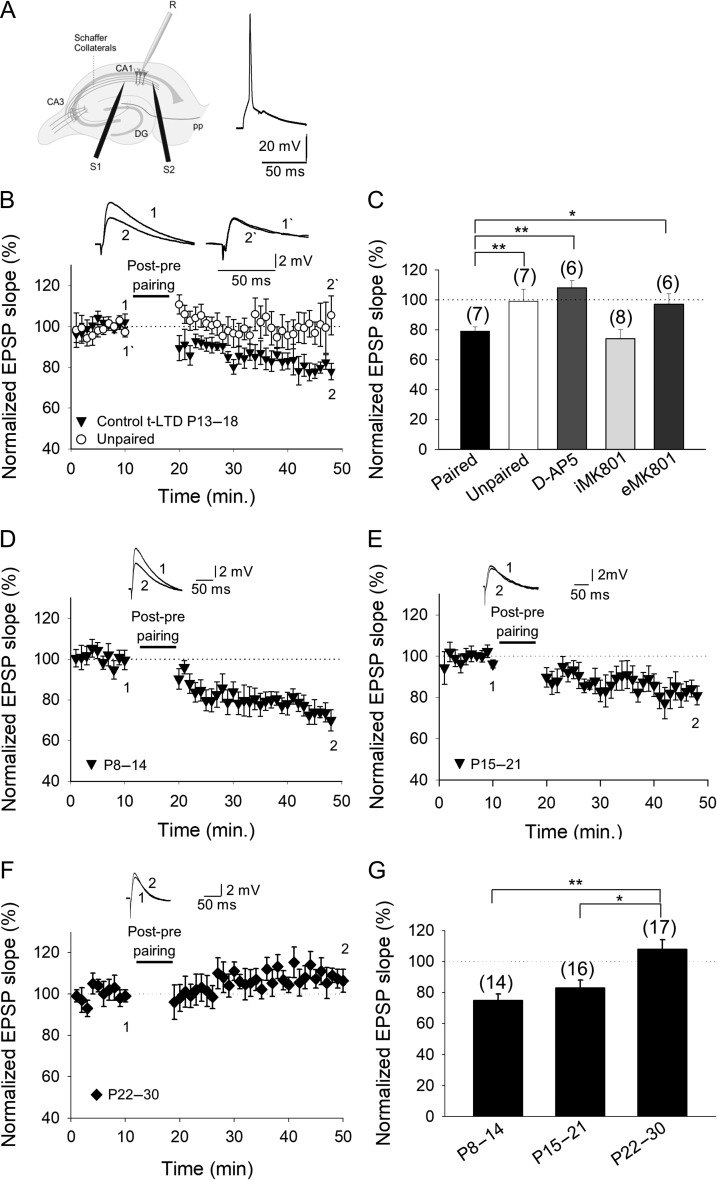
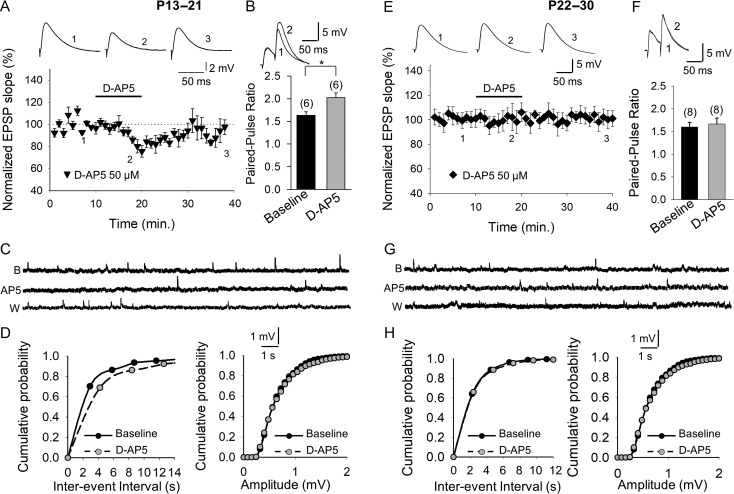
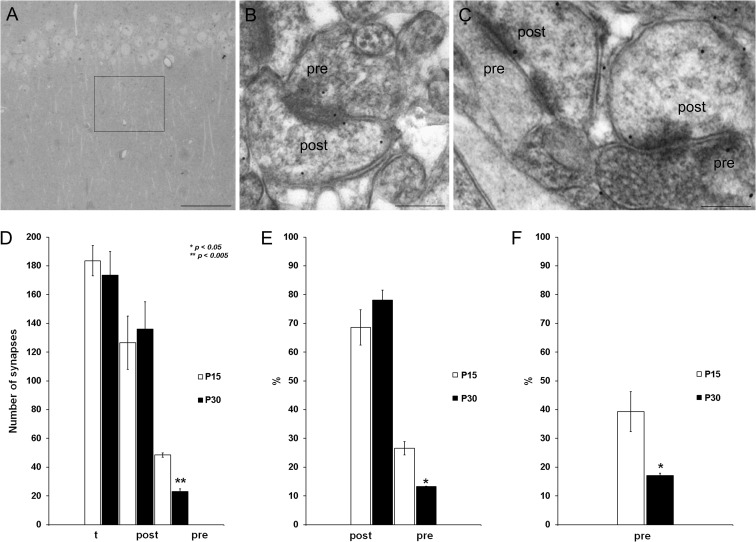
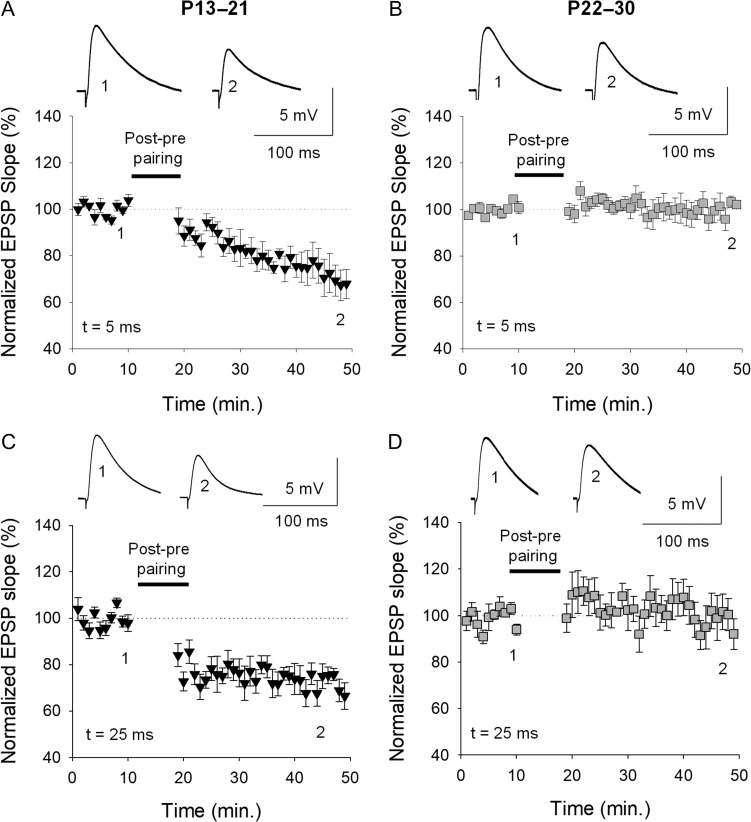
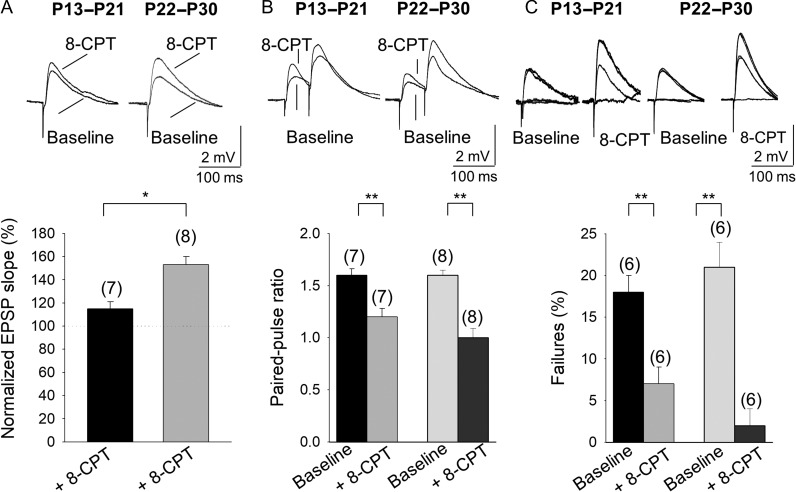
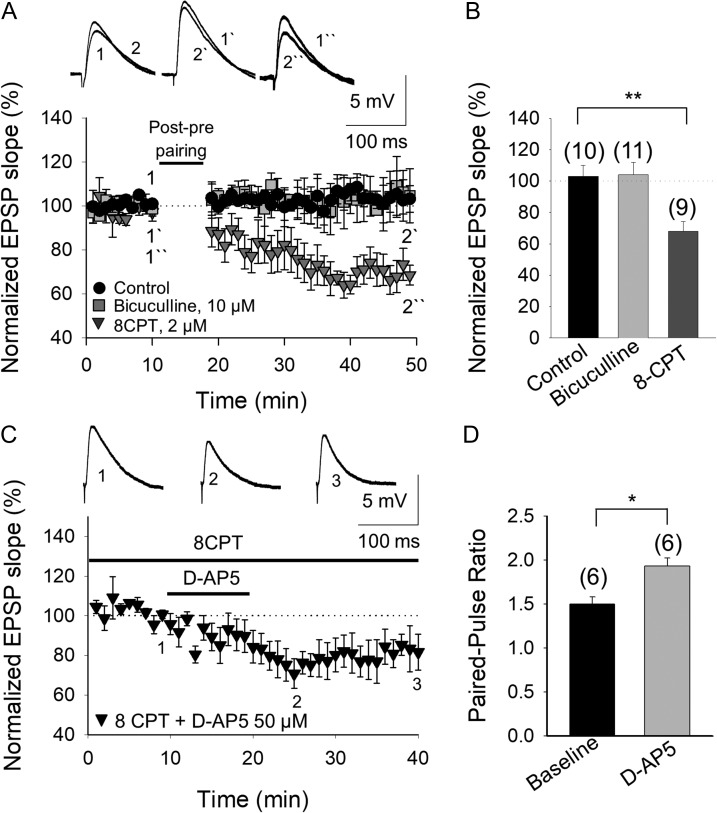
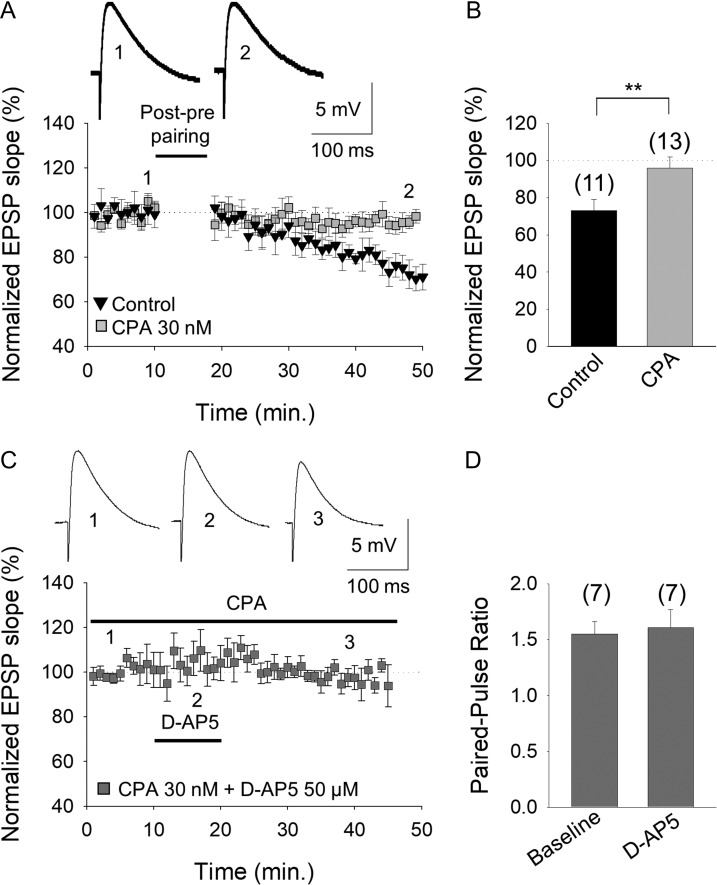
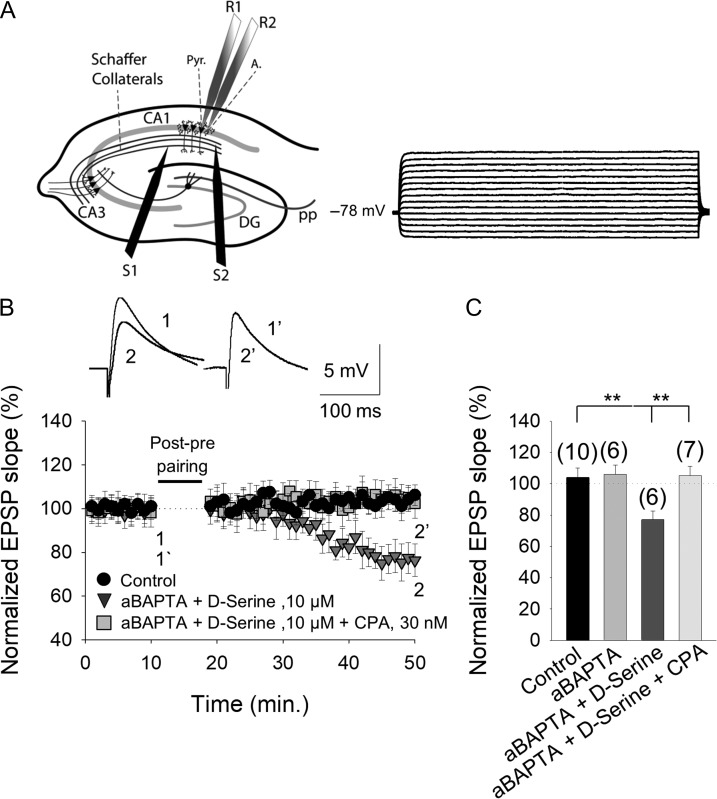
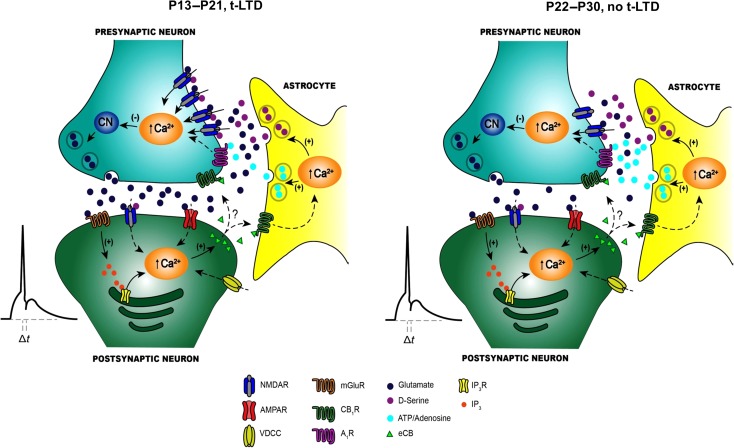
Critical periods of synaptic plasticity facilitate the reordering and refining of neural connections during development, allowing the definitive synaptic circuits responsible for correct adult physiology to be established. Presynaptic spike timing-dependent long-term depression (t-LTD) exists in the hippocampus, which depends on the activation of NMDARs and that probably fulfills a role in synaptic refinement. This t-LTD is present until the third postnatal week in mice, disappearing in the fourth week of postnatal development. We were interested in the mechanisms underlying this maturation related loss of t-LTD and we found that at CA3-CA1 synapses, presynaptic NMDA receptors (pre-NMDARs) are tonically active between P13 and P21, mediating an increase in glutamate release during this critical period of plasticity. Conversely, at the end of this critical period (P22-P30) and coinciding with the loss of t-LTD, these pre-NMDARs are no longer tonically active. Using immunogold electron microscopy, we demonstrated the existence of pre-NMDARs at Schaffer collateral synaptic boutons, where a decrease in the number of pre-NMDARs during development coincides with the loss of both tonic pre-NMDAR activation and t-LTD. Interestingly, this t-LTD can be completely recovered by antagonizing adenosine type 1 receptors (A1R), which also recovers the tonic activation of pre-NMDARs at P22-P30. By contrast, the induction of t-LTD was prevented at P13-P21 by an agonist of A1R, as was tonic pre-NMDAR activation. Furthermore, we found that the adenosine that mediated the loss of t-LTD during the fourth week of development is supplied by astrocytes. These results provide direct evidence for the mechanism that closes the window of plasticity associated with t-LTD, revealing novel events probably involved in synaptic remodeling during development.
Keywords: adenosine receptors; astrocytes; hippocampus; plasticity windows; spike timing-dependent plasticity.
© The Author(s) 2018. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Andrade-Talavera Y, Duque-Feria P, Negrete-Díaz JV, Sihra TS, Flores G, Rodríguez-Moreno A. 2012. Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+-calmodulin at mossy fiber-CA3 synapses. J Neurochem. 122:891–899. - PubMed
-
- Arai A, Kessler M, Lynch G. 1990. The effects of adenosine on the development of long-term potentiation. Neurosci Lett. 119:41–44. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
