

Can Deep Learning Improve Genomic Prediction of Complex Human Traits?
- PMID: 30171033
- PMCID: PMC6218236
- DOI: 10.1534/genetics.118.301298
Can Deep Learning Improve Genomic Prediction of Complex Human Traits?
Abstract
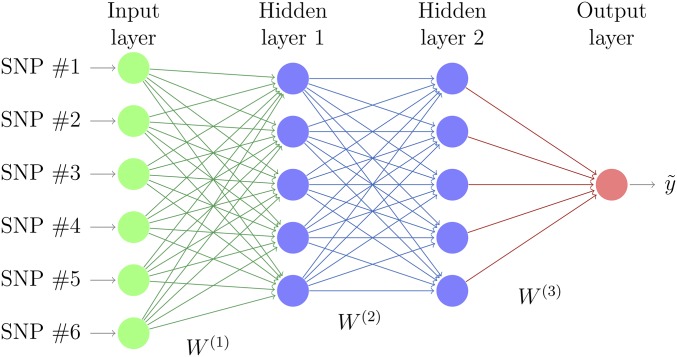
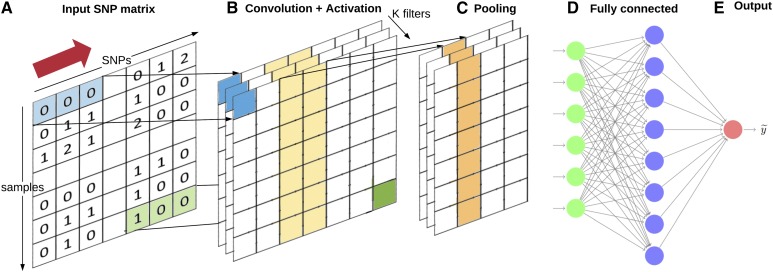
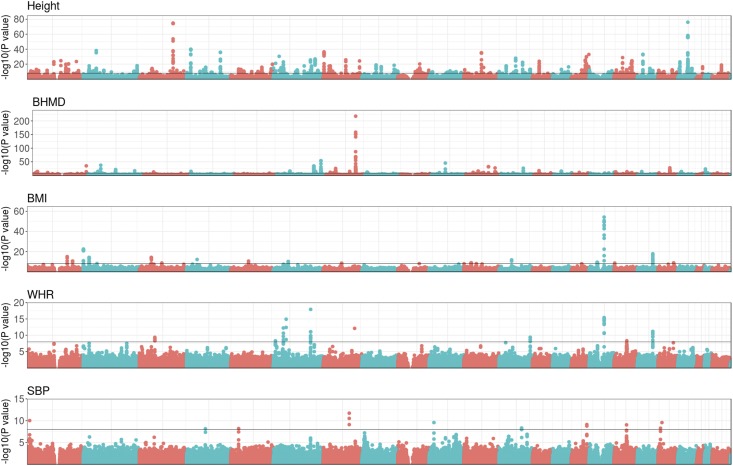
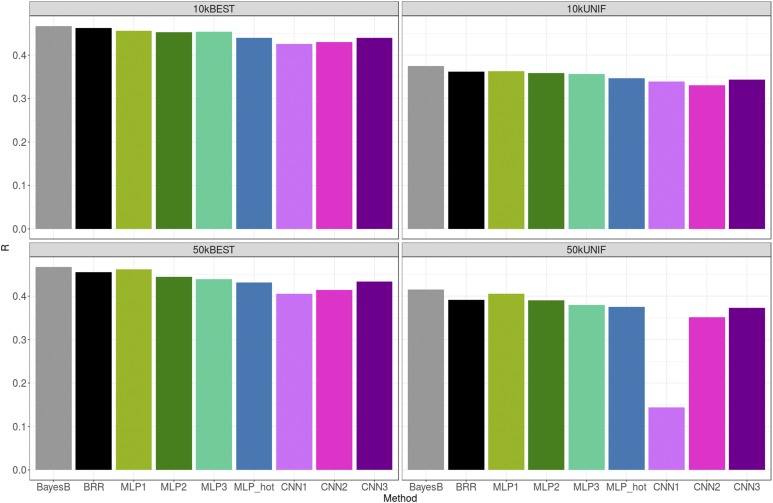
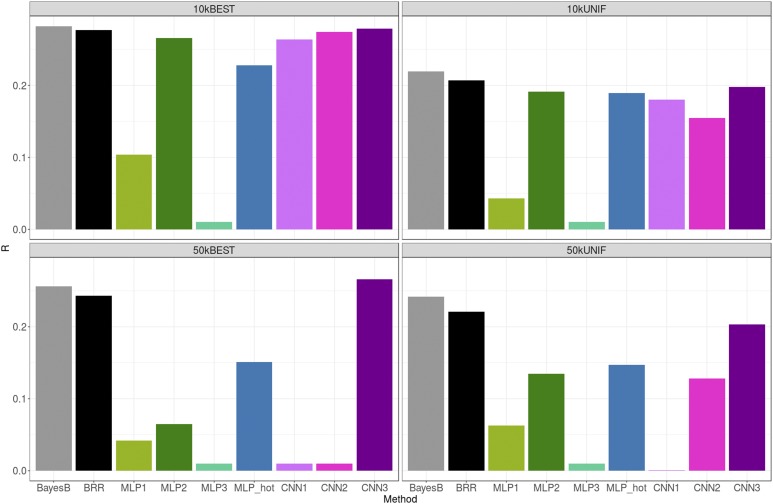
The genetic analysis of complex traits does not escape the current excitement around artificial intelligence, including a renewed interest in "deep learning" (DL) techniques such as Multilayer Perceptrons (MLPs) and Convolutional Neural Networks (CNNs). However, the performance of DL for genomic prediction of complex human traits has not been comprehensively tested. To provide an evaluation of MLPs and CNNs, we used data from distantly related white Caucasian individuals (n ∼100k individuals, m ∼500k SNPs, and k = 1000) of the interim release of the UK Biobank. We analyzed a total of five phenotypes: height, bone heel mineral density, body mass index, systolic blood pressure, and waist-hip ratio, with genomic heritabilities ranging from ∼0.20 to 0.70. After hyperparameter optimization using a genetic algorithm, we considered several configurations, from shallow to deep learners, and compared the predictive performance of MLPs and CNNs with that of Bayesian linear regressions across sets of SNPs (from 10k to 50k) that were preselected using single-marker regression analyses. For height, a highly heritable phenotype, all methods performed similarly, although CNNs were slightly but consistently worse. For the rest of the phenotypes, the performance of some CNNs was comparable or slightly better than linear methods. Performance of MLPs was highly dependent on SNP set and phenotype. In all, over the range of traits evaluated in this study, CNN performance was competitive to linear models, but we did not find any case where DL outperformed the linear model by a sizable margin. We suggest that more research is needed to adapt CNN methodology, originally motivated by image analysis, to genetic-based problems in order for CNNs to be competitive with linear models.
Keywords: Convolutional Neural Networks; GenPred; Genomic Prediction regressions; Multilayer Perceptrons; UK Biobank; complex traits; deep learning; genomic prediction; whole-genome.
Copyright © 2018 by the Genetics Society of America.
Figures
References
-
- Abadi M., Agarwal A., Barham P., Brevdo E., Chen Z., et al. , 2015. TensorFlow: large-scale machine learning on heterogeneous systems. Available at: tensorflow.org. Accessed: July 1, 2018.
-
- Chollet F., 2015. Keras: deep learning library for theano and tensorflow. Available at: https://keras.io/. Accessed May 1, 2018.
-
- de Los Campos, G., and A. Grueneberg, 2017 BGData: a suite of packages for analysis of big genomic data. R package version 1.0.0.9000. Available at:https://github.com/QuantGen/BGData - PMC - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
