

Structural basis of DNA lesion recognition for eukaryotic transcription-coupled nucleotide excision repair
- PMID: 30174298
- PMCID: PMC6340766
- DOI: 10.1016/j.dnarep.2018.08.006
Structural basis of DNA lesion recognition for eukaryotic transcription-coupled nucleotide excision repair
Abstract
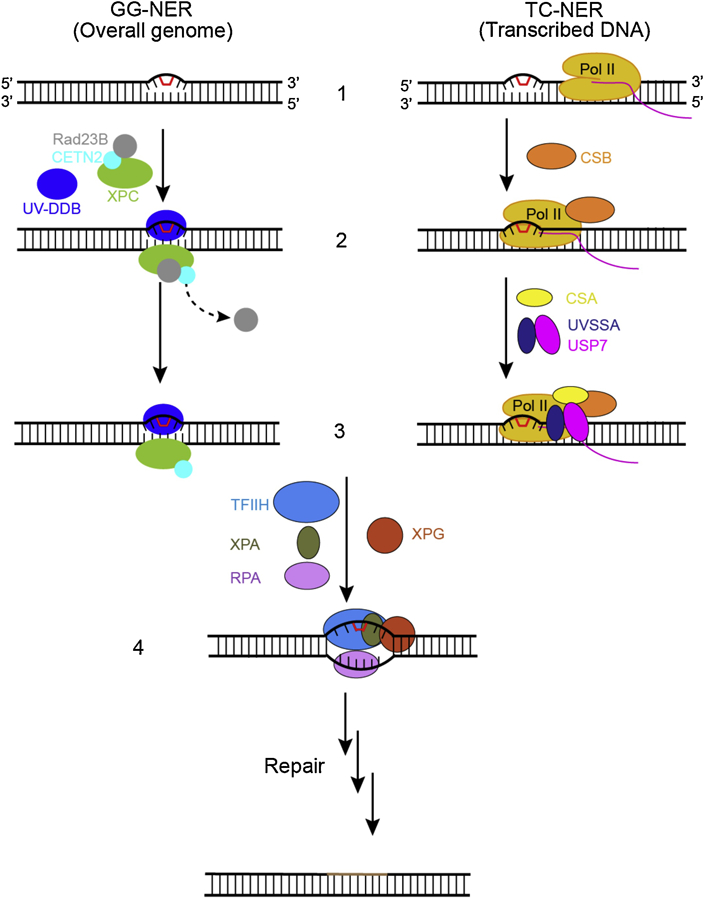
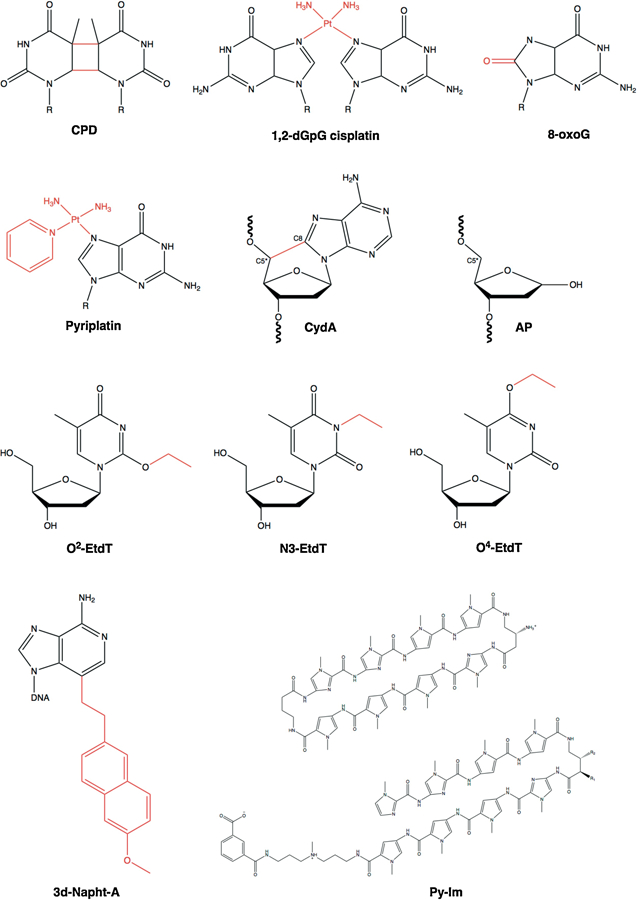
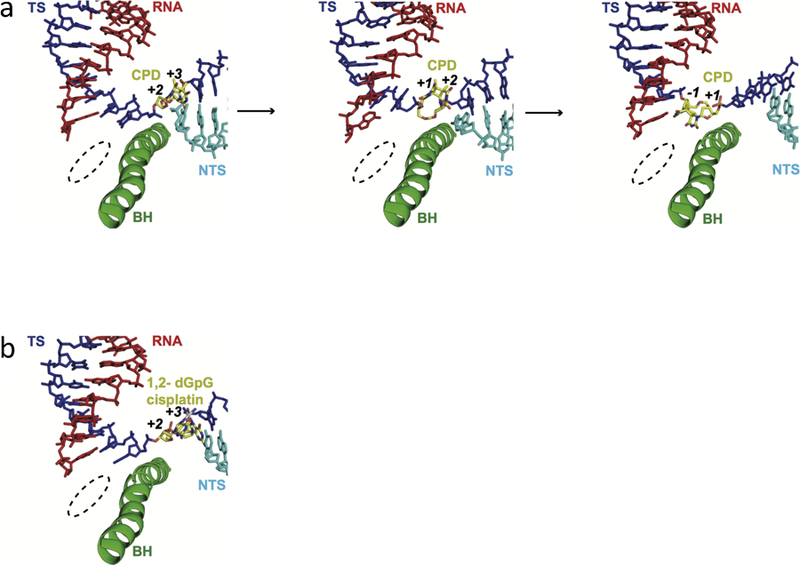
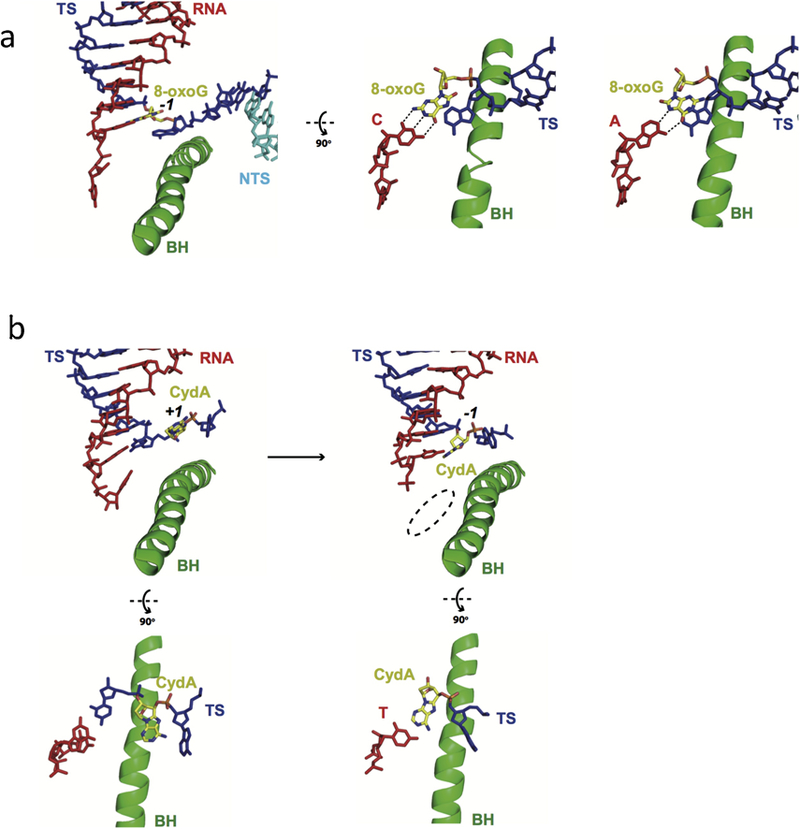
Eukaryotic transcription-coupled nucleotide excision repair (TC-NER) is a pathway that removes DNA lesions capable of blocking RNA polymerase II (Pol II) transcription from the template strand. This process is initiated by lesion-arrested Pol II and the recruitment of Cockayne Syndrome B protein (CSB). In this review, we will focus on the lesion recognition steps of eukaryotic TC-NER and summarize the recent research progress toward understanding the structural basis of Pol II-mediated lesion recognition and Pol II-CSB interactions. We will discuss the roles of CSB in both TC-NER initiation and transcription elongation. Finally, we propose an updated model of tripartite lesion recognition and verification for TC-NER in which CSB ensures Pol II-mediated recognition of DNA lesions for TC-NER.
Keywords: Cockayne syndrome; DNA damage; Lesion recognition; Nucleotide excision repair; RNA polymerase II; Transcription-coupled nucleotide excision repair; Transcriptional arrest.
Copyright © 2018 Elsevier B.V. All rights reserved.
Conflict of interest statement
Conflict of Interest
The authors declare no conflict of interest.
Figures
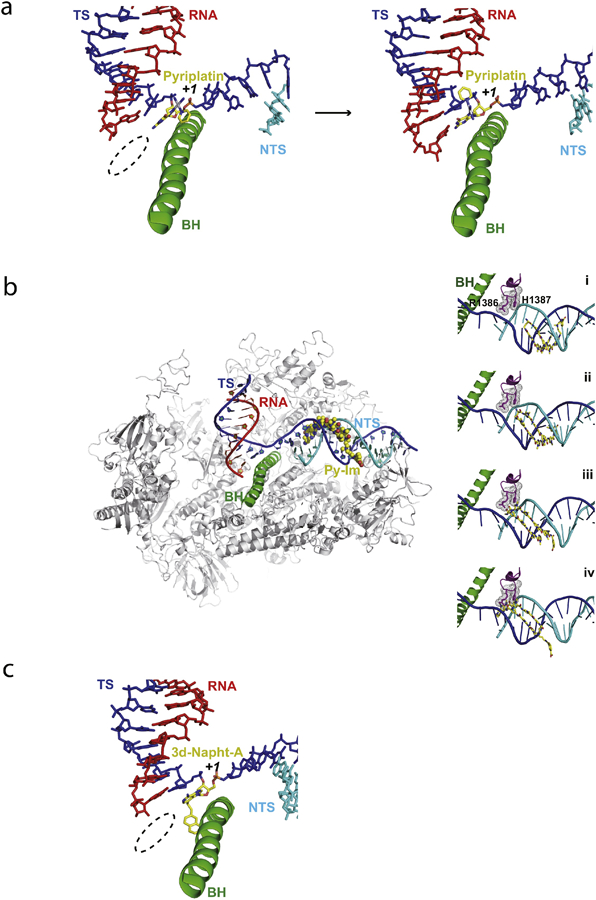
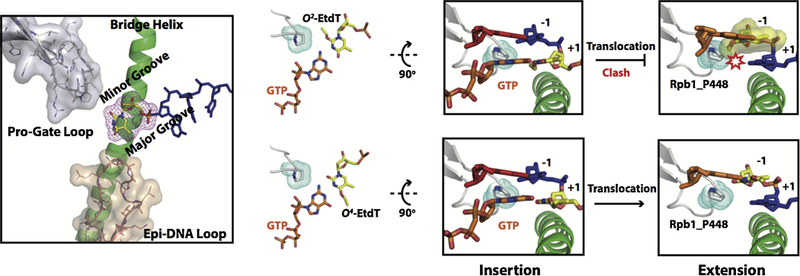
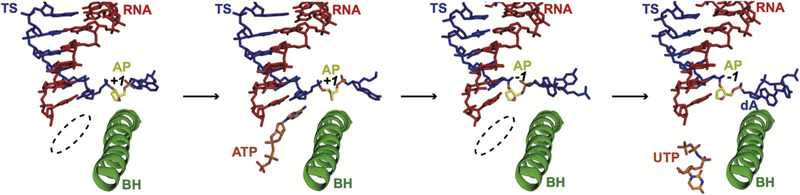
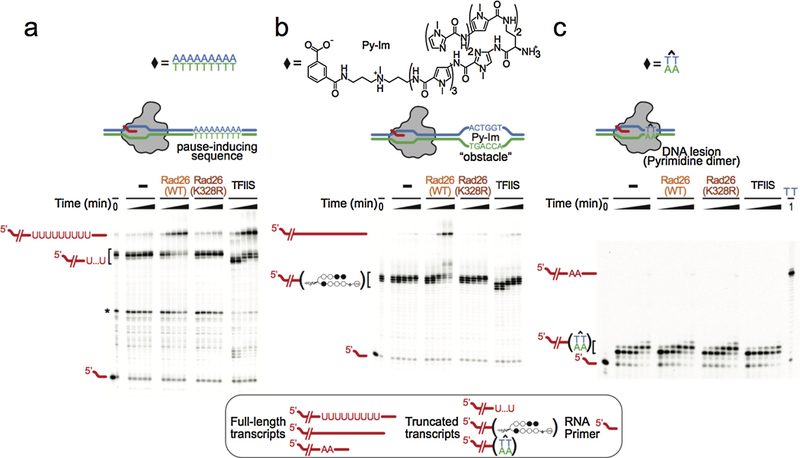

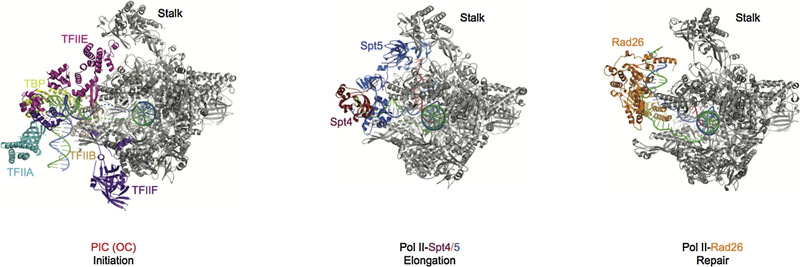
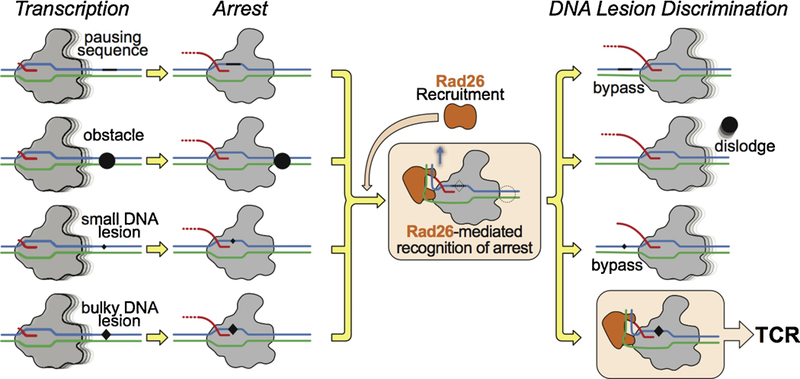
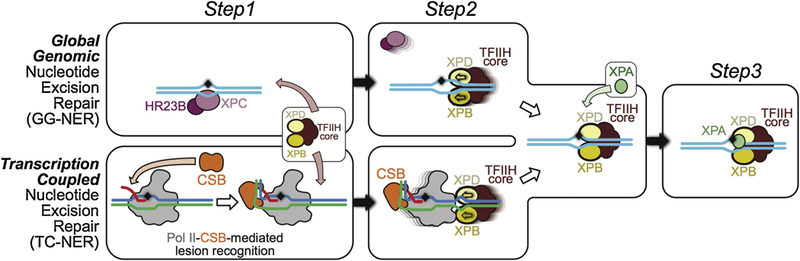
References
-
- Sancar A & Reardon JT (2004) Nucleotide excision repair in E. coli and man. Adv Protein Chem 69:43–71. - PubMed
-
- Hanawalt PC & Spivak G (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9(12):958–970. - PubMed
-
- Bohr VA, Smith CA, Okumoto DS, & Hanawalt PC (1985) DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 40(2):359–369. - PubMed
-
- Mellon I, Spivak G, & Hanawalt PC (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 51(2):241–249. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
