

Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice
- PMID: 30175250
- PMCID: PMC6115194
- DOI: 10.1038/s42003-018-0099-2
Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice
Abstract
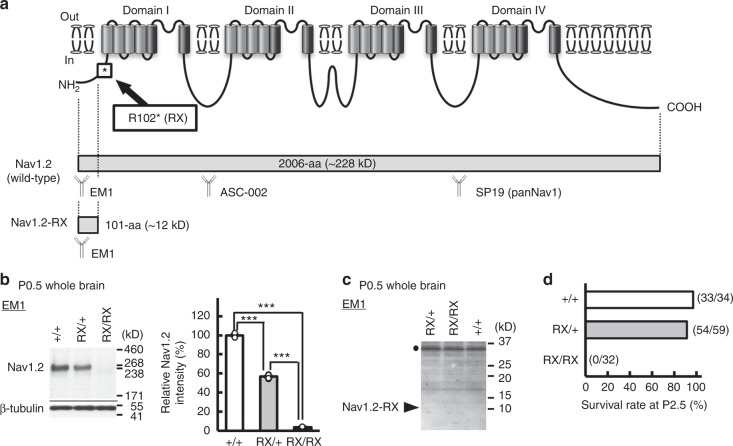
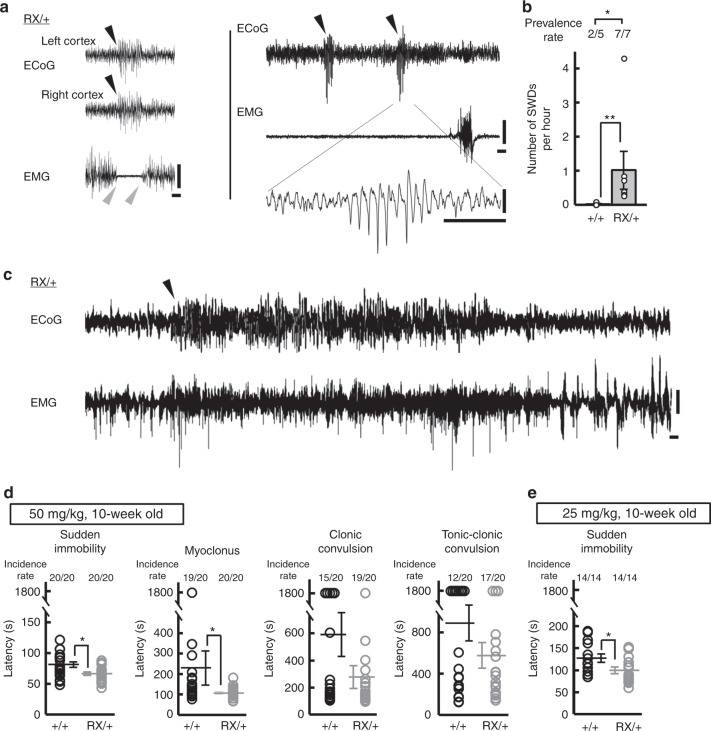
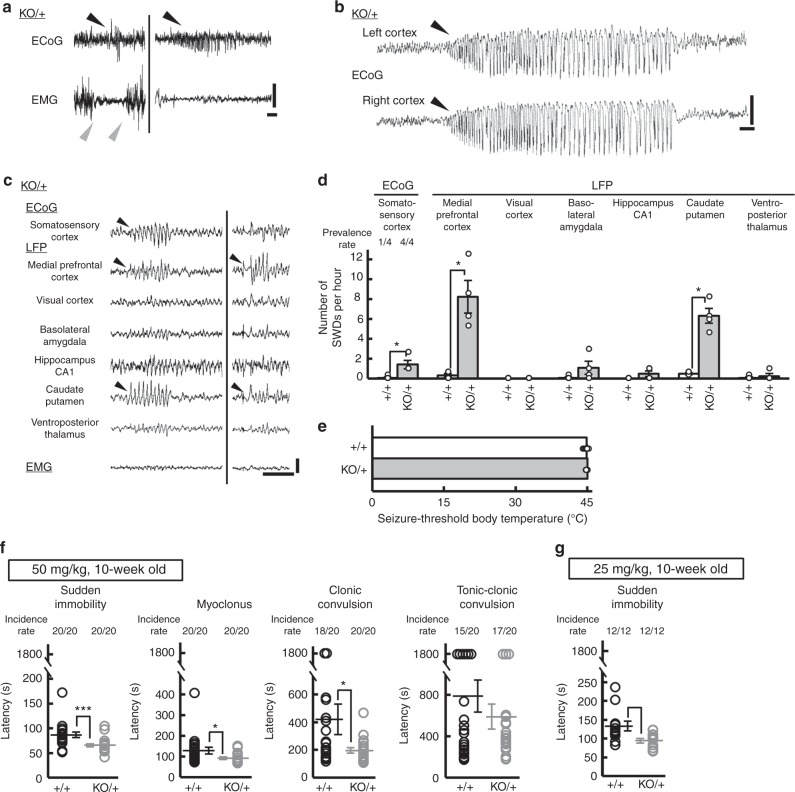
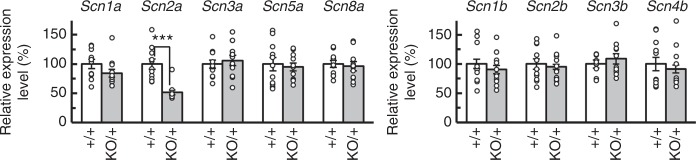
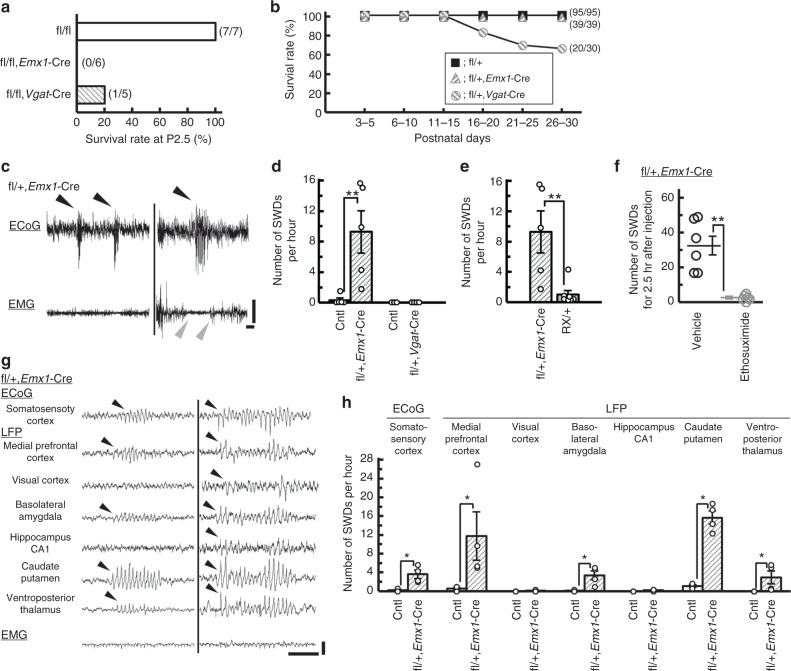
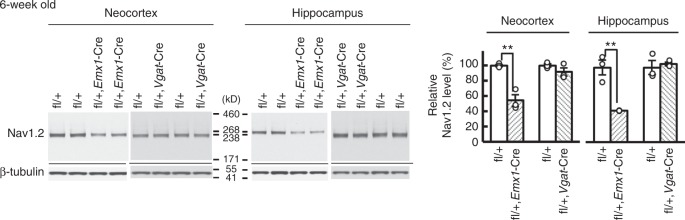
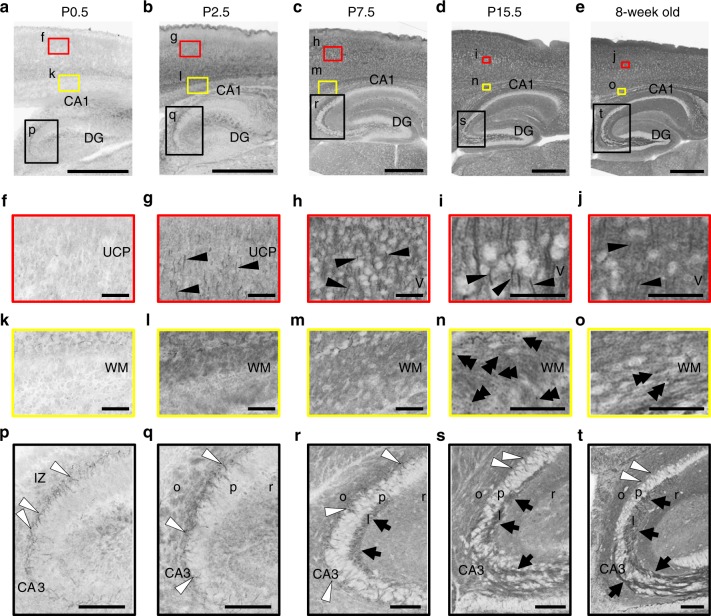
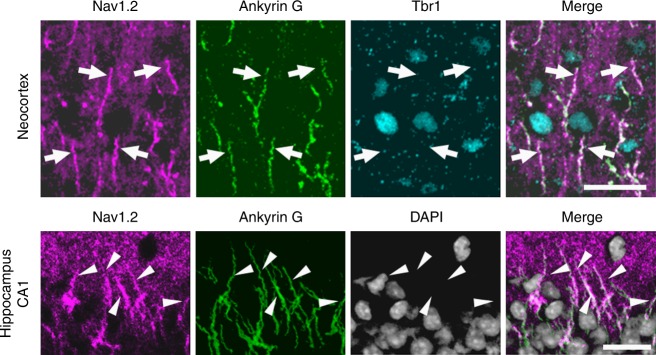
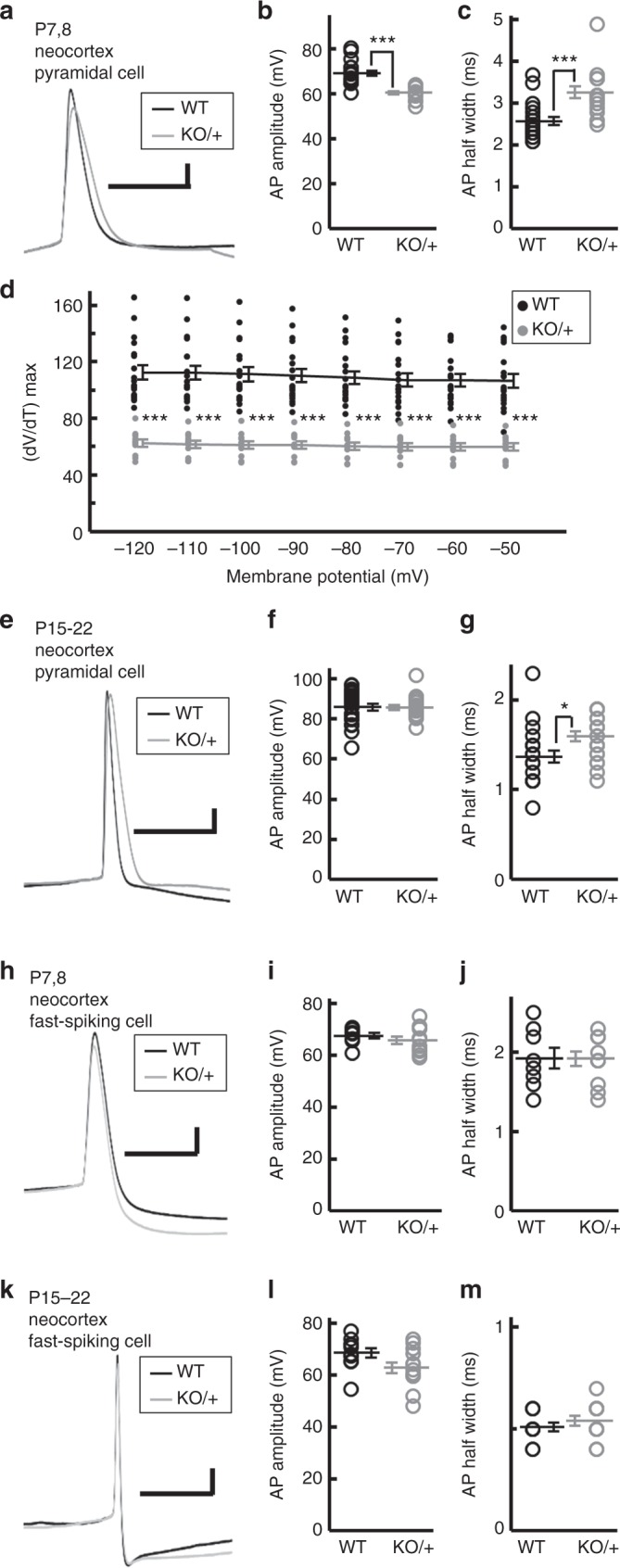
Mutations in the SCN2A gene encoding a voltage-gated sodium channel Nav1.2 are associated with epilepsies, intellectual disability, and autism. SCN2A gain-of-function mutations cause early-onset severe epilepsies, while loss-of-function mutations cause autism with milder and/or later-onset epilepsies. Here we show that both heterozygous Scn2a-knockout and knock-in mice harboring a patient-derived nonsense mutation exhibit ethosuximide-sensitive absence-like seizures associated with spike-and-wave discharges at adult stages. Unexpectedly, identical seizures are reproduced and even more prominent in mice with heterozygous Scn2a deletion specifically in dorsal-telencephalic (e.g., neocortical and hippocampal) excitatory neurons, but are undetected in mice with selective Scn2a deletion in inhibitory neurons. In adult cerebral cortex of wild-type mice, most Nav1.2 is expressed in excitatory neurons with a steady increase and redistribution from proximal (i.e., axon initial segments) to distal axons. These results indicate a pivotal role of Nav1.2 haplodeficiency in excitatory neurons in epilepsies of patients with SCN2A loss-of-function mutations.
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Scn2a severe hypomorphic mutation decreases excitatory synaptic input and causes autism-associated behaviors.JCI Insight. 2021 Aug 9;6(15):e150698. doi: 10.1172/jci.insight.150698. JCI Insight. 2021. PMID: 34156984 Free PMC article.
-
Hyperexcitability in adult mice with severe deficiency in NaV1.2 channels.Int J Physiol Pathophysiol Pharmacol. 2022 Feb 15;14(1):55-59. eCollection 2022. Int J Physiol Pathophysiol Pharmacol. 2022. PMID: 35310859 Free PMC article.
-
Scn2a haploinsufficient mice display a spectrum of phenotypes affecting anxiety, sociability, memory flexibility and ampakine CX516 rescues their hyperactivity.Mol Autism. 2019 Mar 28;10:15. doi: 10.1186/s13229-019-0265-5. eCollection 2019. Mol Autism. 2019. PMID: 30962870 Free PMC article.
-
Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities.Epilepsia. 2014 Apr;55(4):e25-9. doi: 10.1111/epi.12554. Epub 2014 Mar 1. Epilepsia. 2014. PMID: 24579881 Review.
-
SCN2A channelopathies: Mechanisms and models.Epilepsia. 2019 Dec;60 Suppl 3:S68-S76. doi: 10.1111/epi.14731. Epilepsia. 2019. PMID: 31904120 Review.
Cited by
-
Sodium channelopathies of skeletal muscle and brain.Physiol Rev. 2021 Oct 1;101(4):1633-1689. doi: 10.1152/physrev.00025.2020. Epub 2021 Mar 26. Physiol Rev. 2021. PMID: 33769100 Free PMC article. Review.
-
Venom Peptides with Dual Modulatory Activity on the Voltage-Gated Sodium Channel NaV1.1 Provide Novel Leads for Development of Antiepileptic Drugs.ACS Pharmacol Transl Sci. 2019 Nov 25;3(1):119-134. doi: 10.1021/acsptsci.9b00079. eCollection 2020 Feb 14. ACS Pharmacol Transl Sci. 2019. PMID: 32259093 Free PMC article.
-
The voltage-gated sodium channel inhibitor, 4,9-anhydrotetrodotoxin, blocks human Nav1.1 in addition to Nav1.6.Neurosci Lett. 2020 Apr 17;724:134853. doi: 10.1016/j.neulet.2020.134853. Epub 2020 Feb 27. Neurosci Lett. 2020. PMID: 32114117 Free PMC article.
-
Microglial over-pruning of synapses during development in autism-associated SCN2A-deficient mice and human cerebral organoids.Mol Psychiatry. 2024 Aug;29(8):2424-2437. doi: 10.1038/s41380-024-02518-4. Epub 2024 Mar 18. Mol Psychiatry. 2024. PMID: 38499656
-
Scn2a Haploinsufficiency in Mice Suppresses Hippocampal Neuronal Excitability, Excitatory Synaptic Drive, and Long-Term Potentiation, and Spatial Learning and Memory.Front Mol Neurosci. 2019 Jun 4;12:145. doi: 10.3389/fnmol.2019.00145. eCollection 2019. Front Mol Neurosci. 2019. PMID: 31249508 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
