

Insights into the genotype-phenotype correlation and molecular function of SLC25A46
- PMID: 30178502
- PMCID: PMC6240357
- DOI: 10.1002/humu.23639
Insights into the genotype-phenotype correlation and molecular function of SLC25A46
Abstract
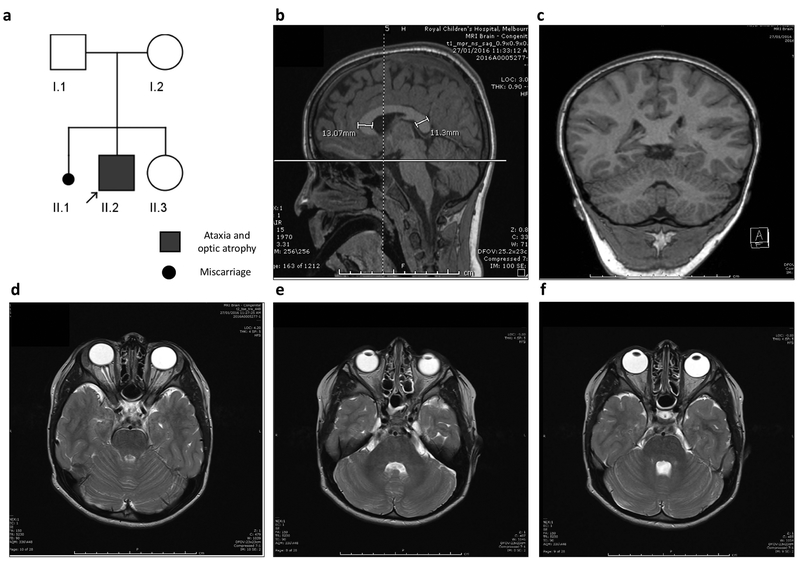
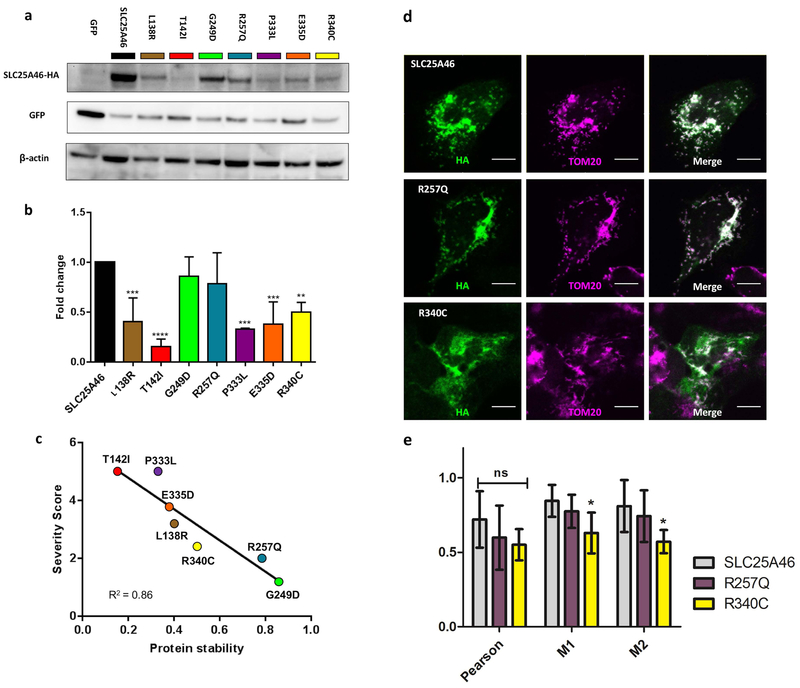
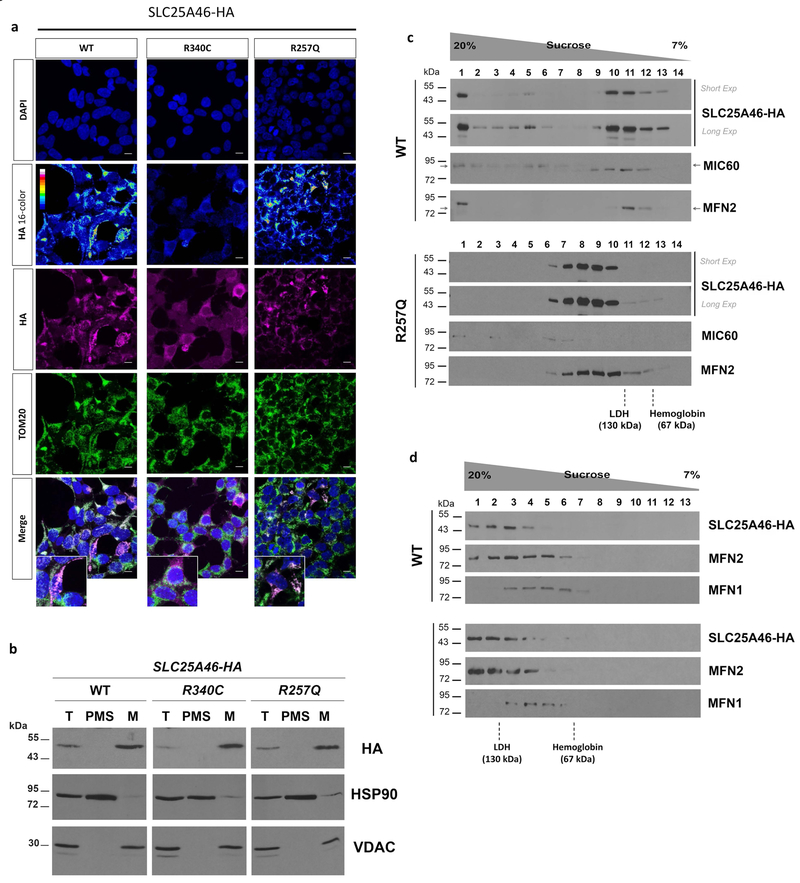
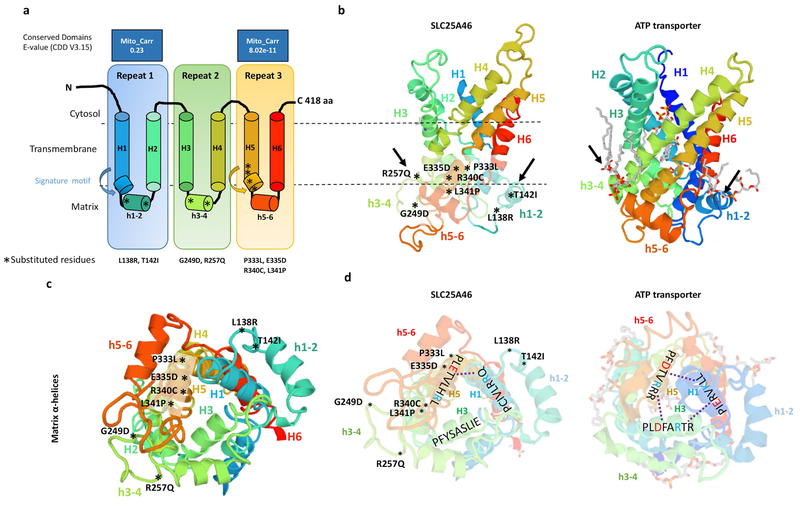
Recessive SLC25A46 mutations cause a spectrum of neurodegenerative disorders with optic atrophy as a core feature. We report a patient with optic atrophy, peripheral neuropathy, ataxia, but not cerebellar atrophy, who is on the mildest end of the phenotypic spectrum. By studying seven different nontruncating mutations, we found that the stability of the SLC25A46 protein inversely correlates with the severity of the disease and the patient's variant does not markedly destabilize the protein. SLC25A46 belongs to the mitochondrial transporter family, but it is not known to have transport function. Apart from this possible function, SLC25A46 forms molecular complexes with proteins involved in mitochondrial dynamics and cristae remodeling. We demonstrate that the patient's mutation directly affects the SLC25A46 interaction with MIC60. Furthermore, we mapped all of the reported substitutions in the protein onto a 3D model and found that half of them fall outside of the signature carrier motifs associated with transport function. We thus suggest that there are two distinct molecular mechanisms in SLC25A46-associated pathogenesis, one that destabilizes the protein while the other alters the molecular interactions of the protein. These results have the potential to inform clinical prognosis of such patients and indicate a pathway to drug target development.
Keywords: Ataxia; Mitochondria; Optic atrophy; SLC25A46.
© 2018 Wiley Periodicals, Inc.
Conflict of interest statement
Conflict of Interest Statement
The Authors have no conflicts of interest to disclose.
Figures

References
-
- Abrams AJ, Hufnagel RB, Rebelo A, Zanna C, Patel N, Gonzalez MA, … Dallman JE (2015). Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat Genet, 47(8), 926–932. doi: 10.1038/ng.3354 - DOI - PMC - PubMed
-
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, … Wissinger B (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet, 26(2), 211–215. doi: 10.1038/79944 - DOI - PubMed
-
- Charlesworth G, Balint B, Mencacci NE, Carr L, Wood NW, & Bhatia KP (2016). SLC25A46 mutations underlie progressive myoclonic ataxia with optic atrophy and neuropathy. Mov Disord, 31(8), 1249–1251. doi: 10.1002/mds.26716 - DOI - PubMed
-
- Chen H, McCaffery JM, & Chan DC (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell, 130(3), 548–562. doi: 10.1016/j.cell.2007.06.026 - DOI - PubMed
-
- Coonrod EM, Karren MA, & Shaw JM (2007). Ugo1p is a multipass transmembrane protein with a single carrier domain required for mitochondrial fusion. Traffic, 8(5), 500–511. doi: 10.1111/j.1600-0854.2007.00550.x - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
